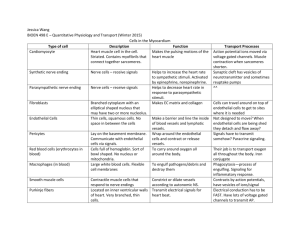
Open Access version via Utrecht University Repository
advertisement

Immunological and fibrotic mechanisms in Cardiac Allograft Vasculopathy Master thesis Biology of Disease Manon Jansen 22-7-2014 Supervisors: Manon Huibers, MSc Roel de Weger, PhD Second reviewer: Henny Otten, PhD Abstract Cardiac allograft vasculopathy (CAV) has a high prevalence among patients who received heart transplantation (HTx). CAV is a multiple factorial process in which the immune system plays a large role. In this thesis, the data on the immunological and fibrotic processes that are involved in the development of CAV are summarized. During the pathogenesis of CAV, cells from the innate and the adaptive immune system cooperate to reject the foreign organ that is transplanted. The inflammatory response results in dysfunction of the endothelium and migration and proliferation of smooth muscle cells (SMCs). Apoptosis and factors secreted by both the endothelium as the SMCs lead to fibrosis. The migration of SMCs together with fibrosis provoke concentric intimal thickening of the coronary arteries, which is the main characteristic of CAV. Laymen’s summary When patients receive a heart transplant, it is often rejected after several years. One of the major causes for this rejection is cardiac allograft vasculopathy (CAV). CAV is a disease in which the arteries that surround the transplanted heart are beginning to thicken. This results in narrow arteries and can, as a result, lead to reduced oxygen supply, resulting in a heart attack. Patients develop CAV because the immune system of the patient recognizes the transplanted heart as foreign and attacks it. This is comparable to how the body attacks substances that cause disease such as viruses. Because the immune system is activated, other cells in the arteries surrounding the heart are also activated. This can lead to death of these cells but also to production of growth factors which stimulate the growth of cells in the arterial wall which leads to thickening of the arterial wall. Introduction Worldwide more than 5,000 heart transplants are performed each year1. Heart transplantation is implemented in patients who are concerned with end stage heart failure. Unfortunately, there is a huge shortage of donor organs. Recent numbers showed that in 2013, 45,000 patients were on the waiting list who could not be helped, which limits the number of heart transplants 2. Although the survival rates after heart transplantation have increased enormously in the last decade, cardiac allografts still fail in 30% of the cases within 10 years after transplantation3. The one year survival rate however, is about 90%. The reason for this is that short-term survival has improved by immunosuppressive therapy and improved surgical techniques. But the long-term survival had no such improvements in the past years. The long-term survival of a heart can be comprised by several factors. One of the main causes for graft failure in general is rejection. An allograft can be rejected in various ways: via acute rejection, antibody-mediated rejection (AMR) or chronic rejection. Acute rejection takes place several days after transplantation, when the immune system recognizes the transplanted organ as foreign and attacks it. For acute rejection, Margareth Billingham described already in 1990 a four-stage classification for cellular heart transplant rejection. This first classification is nowadays known as Billingham’s criteria or Stanford classification. The Stanford classification describes the four stages of CAV as follows. If there is no rejection present, the grade is ‘Absent’. If there is focal or diffuse infiltration of lymphocytes without myocyte damage, the grade is ‘Mild’. The grade is ‘Moderate’ if these lymphocytic infiltrates do damage the myocytes. The final grade is ‘Severe’ in which the infiltration of lymphocytes is associated with neutrophils and interstitial hemorrhage. After several 2 years of research, this classification was refined in 2005 by the International society for heart lung transplantation (ISHLT)4. The grading system is as follows: Grade 0 R – no rejection; Grade 1 R, mild – interstitial and/or perivascular infiltrate with focal points of myocyte damage; Grade 2 R, moderate – two or more foci of infiltrate with associated myocyte damage; and Grade 3 R, severe– diffuse infiltrate with multifocal myocyte damage and possibly edema, hemorrhage, and vasculitis4. Antibody mediated rejection was not included in these criteria, although the presence can be established with AMR 0 (AMR is absent), AMR 1 (AMR is present in histologic findings or immunopathologic findings), AMR 2 (AMR is pathologic) or AMR 3 (AMR is severe pathologic) 5. In case the allograft is rejected in an antibody-mediated way, antibodies targeting human leukocyte antigen (HLA) molecules, non-HLA molecules such as vimentin6 and donor endothelial cells are present in the host’s circulation7. In case the antibodies are already preformed and present in the blood of the host before transplantation, the allograft rejection takes place within minutes after transplantation. This is called hyperacute rejection. Chronic rejection occurs a few months or even years after transplantation. Chronic rejection is a process in which multiple factors play a role. Not only the immune system but also processes like vascular remodeling and fibrosis. In cardiac allografts, chronic rejection in the form of cardiac allograft vasculopathy (CAV) has a high prevalence. Thirty percent of the patients have CAV five years after transplantation4. CAV is a pathological process that affects the vasculature of the transplanted heart. It is characterized by concentric thickening of the blood vessel wall, which is due to proliferation of smooth muscle cells (SMC) and intimal hyperplasia in the coronary vessels and the intramyocardial microvasculature. This leads to narrowing of the vessel and subsequently to ischemia in the graft8. CAV is difficult to diagnose. In the early stages patients are often asymptomatic, while in later stages patients can develop silent myocardial ischemia or even suddenly die. Undetected CAV may also present in heart failure due to global ischemia. Pathologically, the severity of CAV can be identified (Huibers et al. 2014, accepted Atheroscleriosis). There are several risk factors that are correlated with CAV. These risk factors can be transplant associated or transplant non-associated. Also there are risk factors that are due to complications during or after the transplantation (e.g. infections). Examples of transplant associated risk factors are HLA-mismatch, the time the organ donor was brain death and donor age9. Risk factors that are not directly linked to the transplant are hypertension, hyperlipidemia and diabetes4, 10. CAV versus atherosclerosis CAV is often compared to atherosclerosis. Some people even refer to CAV as graft arteriosclerosis11. To avoid confusion, the ISHLT formulated a standard nomenclature for CAV8. These guidelines also involve a histological grading system to distinguish CAV from atherosclerosis. Although there are similarities between CAV and atherosclerosis, there are several aspects that are specific for CAV. CAV is a diffuse process in which the total surface of smaller vessels is homogenously affected, while atherosclerosis is present in eccentric lesions in medium- and large-sized arteries. Furthermore, CAV has a rapid onset; it can develop within a few months and the development is similar in males, females and pediatric patients, whereas atherosclerosis takes years to evolve8. Looking histological to CAV, the differences between atherosclerosis and CAV can also be clearly seen. In healthy individuals, the coronary vessels are roughly composed of the endothelial layer, the tunica intima, tunica media and tunica adventitia. The tunica intima lies between the endothelium and the lamina 3 elastic interna and consists mainly of SMCs12. In patients with CAV, the neointima consists of two layers. One layer is linked to the lamina elastic interna of the neointima but the second layer is not composed of SMCs, like in healthy people, but of loose connective tissue12. Atherosclerosis can be seen histological as focal lesions which are more asymmetrical than CAV lesions. These lesions can contain a lipid core and calcification but not in all cases. The lamina elastic interna is interrupted within an atherosclerotic plaque, while in CAV the elastica interna is intact. Figure 1. Similarities and differences between atherosclerosis and allograft vasculopathy Arteries that are affected by atherosclerosis contain focal lesions while vessels that are affected by allograft vasculopathy are narrowed in a diffuse way. Not only the location of the lesions differs between atherosclerosis and allograft vasculopathy, also the composition of the lesions is different. An atherosclerotic plaque often contains a lipid core and calcification, while in an allograft vasculopathy affected vessel the intima is thickened and contains loose connective tissue instead of smooth muscle cells. Colvin-Adams et al. 2013 So, there are many differences between atherosclerosis and CAV. In atherosclerosis research, more and more knowledge is gained about the extensive role of the immune system in the pathogenesis of atherosclerosis. But what causes the vascular wall changes in CAV? The currently leading theory states that the development of a CAV lesion is, just like in other coronary vascular diseases, preceded by endothelial dysfunction13. In this review, we will present an overview of the main currently leading theories about the pathogenesis of CAV. The immunological mechanisms that may lead to fibrosis, but also causes and pathological changes that are part of CAV will be discussed. 4 Chapter 1: What are the main causes for CAV? There are various theories about the causes of CAV. These causes have a variety of characteristics and differ from immunological to non-immunological. Several reviews already described important risk factors for developing CAV10, 14-16. Therefore, we will discuss in this chapter the most relevant causes for understanding the development of CAV in an immunological and fibrotic perspective. Various general cardiovascular risk factors are also being described as important for CAV: hyperlipidemia, diabetes mellitus, smoking, hypertension, body weight and age10, 14, 15. Next to these factors, there are also less known risk factors for CAV such as viral infections, ischemia-reperfusion injury (I-R injury) and brain death of the donor 17. Cytomegalovirus Viral infections have been correlated with accelerated CAV. The cytomegalovirus (CMV) is the best known in this relation18. Opportunistic infections often occur in patients who receive immunosuppressive drugs. CMV is one of the major opportunistic pathogens in patients after HTx. CMV is a virus that invades several cell types that are involved in CAV: SMCs, fibroblasts, macrophages and endothelial cells19. By invading the endothelial cells in the arteries, a pro-CAV state can be created. This pro-CAV state is established in various ways. CMV infection contributes not only by activation of the host’s immune system but also by upregulation of adhesion molecules on the endothelial cells, and promoting the adhesion, activation and transendothelial migration of leukocytes20. Furthermore, CMV inhibits the NO synthase pathway in endothelial cells and increases cytokine production of several cell types, especially production of IFN-γ is stimulated21. That CMV is involved in CAV is shown by Potena et al. They treated two groups of patients with CAV. One group received prophylaxis to fight CMV infection, the other group did not receive this treatment. Potena and colleagues found that the patients with prophylaxis treatment had a delayed onset of CMV infection; CAV was reduced in these patients21. Next to the immunological trigger, CMV is able to reduce apoptosis in SMCs because it encodes two proteins that block the apoptotic pathway. By initiating endothelial cells to produce growth factors such as platelet derived growth factor (PDGF), SMC proliferation is stimulated. This stimulation of growth factors and reduction in apoptosis could contribute to the SMC proliferation and intimal thickening (Chapter 3, The role of smooth muscle cells in fibrosis). Ischemia reperfusion injury Transplantation of a solid organ is often accompanied by ischemia-reperfusion injury (I-R injury)22. During ischemia, the blood supply is interrupted which leads to anaerobic metabolism and loss of transport function of cells15. During reperfusion, reactive oxygen species are released that cause injury to the allograft but also upregulate adhesion molecules on endothelial cells15. This is followed by release of stress proteins such as heat shock proteins and uric acid17, 22. These stress proteins can trigger the innate immune system via toll like receptors (TLRs). The activation of TLRs in turn can lead to both innate and adaptive immune responses (Chapter 2, Innate immunity, Toll like receptors). For a heart allograft, it is also extremely important to limit the time of ischemia. As a heart is taken out of the donor, the so called cold ischemia starts. Research already indicated that extended cold ischemia has a negative impact on the quality of the graft23. Devitt et al. showed that the formation 5 of a CAV lesion begins earlier when cold ischemia is prolonged. They demonstrate that this might be due to an increased neutrophil and CD8+ T cell influx and more proliferation of SMCs23. This increased influx of cells can be explained by the increase of adhesion molecules on endothelial cells during reperfusion. HLA mismatch The risk of CAV increases as the number of HLA mismatches but also the number and duration of cellular rejection episodes increase10, 24. If two or more rejection episodes occur, the prevalence of CAV is significantly increased. Research even shows that the risk of rejection is 25% elevated if the recipient has two HLA mismatches with the donor compared to null or one mismatch25. It is also been demonstrated that a HLA mismatch predicts acute rejection resulting in in-hospital mortality after HTx. This might be due to the fact that HLA matching is not always taken into account for HTx patients. The reason for this is that these patients need to be operated as soon as possible and cannot wait for a heart that has better HLA matching25. Mismatched HLA molecules trigger the alloimmune response (Chapter 2, The immune system). Donor factors The quality of the donor heart is important for patients receiving HTx. Several donor factors can influence the quality of the graft but also the development of CAV. Brain death Research indicates that brain death of the donor can impact the heart allograft and with that the recipient’s prognosis26. Several theories exist to explain why brain death affects the donor heart. One of these theories is that brain death activates IL-6 receptors. IL-6 is correlated with impaired cardiac allograft function (Chapter 2, The immune system). This activation may lead to allograft damage and in time to CAV26. Another theory indicates that brain death induces a calcium overflow injury that affects coronary SMCs and cardiomyocytes, resulting in less efficient contraction of the heart. Furthermore, brain death elevates matrix metalloproteinases (MMPs) and pro-apoptotic factors which can contribute to the development of CAV (Chapter 2, Innate immunity, macrophages; Chapter 3, Fibrosis)24, 26. Although donor age seems to be important in solid organ transplantations, in HTx patients no correlation was found between the age of the donor and the severity of CAV27. Donor specific antibodies Donor specific antibodies (DSA) are an important risk factor for CAV. DSA are antibodies that are reactive with human leukocyte antigen (HLA) molecules of the donor. Graft endothelial cells express HLA in high amounts, which causes the DSA to target these cells. By accumulating on the graft endothelial cells, the complement system is activated28(Chapter 2, The immune system, complement system). 6 General Cardiovascular Smoking Body weight (BMI>30) Hypertension Hyperlipidemia Diabetes mellitus Transplant associated Ischemia-reperfusion injury/cold ischemia CMV infection HLA mismatch Brain death donor Donor specific antibodies Table 1. Risk factors for cardiac allograft vasculopathy Risk factors for CAV are divided into general cardiovascular risk factors and transplant associated risk factors. Another division that is made is immunological versus non-immunological risk factors. 7 Chapter 2: The immune system The general theory is that CAV is mainly an alloimmune process. An indication for this theory is that CAV only affects donor arteries and not recipient arteries. New evidence to support this approach is generated by a retrospective study by Guihaire et al. They show that patients who receive a heartlung transplantation (HLTx) develop significantly less severe CAV compared to HTx patients. The suggested hypothesis states that this effect is due to immune tolerance also known as ‘the combieffect’29. This means that the lymphoid tissue that is associated with the lungs concentrates the immune reaction in the lungs. This leads to less immune activation in the heart and also less CAV. This combi-effect is not only seen in HLTx but also in heart-kidney transplantation29. This study indicates that there is a link between the activity of immune responses and the development of CAV. But which cells are mostly responsible for the development of CAV? The human immune system contains a variety of cells. After transplantation of a solid organ, the recipient’s immune system recognizes the graft as foreign and attacks it. In this process there are many different immune cells involved. The main reason why a graft is rejected is because there are major antigenic differences between a donor and a host. The major histocompatibility complexes (MHC), or in humans the human leukocyte antigen (HLA), in all individuals are different2. By recognition of the foreign HLA, cellular and humoral immunity is initiated. The cellular immunity can be activated via three pathways: the direct, the indirect and the semi-direct pathway. In the direct pathway, the MHC molecules on the donor cell surface are in total recognized by antigen presenting cells (APCs) of the host. When the host T cells encounter the donor APCs, they recognize the foreign antigen-MHC complex on the host APCs and become activated. Furthermore, the host T cells can directly recognize donor MHC molecules2. The activation of the T cells takes place in the transplanted heart or after the APCs migrated to the draining lymph nodes30. In the indirect pathway, the host APCs process donor MHC and present them as antigens to host T cells. This process is similar to the processing and presentation of other foreign antigens like bacteria. The semi-direct pathway involves host APCs that acquire donor MHC via cell-cell contact. This activates the host T cells2, 17. The current idea is that the direct pathway is mostly responsible for acute rejection and the indirect and semidirect pathway for chronic rejection 2. However, also other components of the immune system contribute to the rejection of an allograft. Because less knowledge is gained about the innate contributors compared to the mechanisms that involve cells from the adaptive immune system, the adaptive immune system will be discussed first. Adaptive immunity CD4+ and CD8+ T cells To establish which types of cells are present in the lesions, van Loosdregt et al. obtained hearts from HTx patients and checked which mononuclear cells were present. They found that T cells were the most abundant type of cell in the CAV lesions. From these T cells, the CD4+ T cells were twice as much present as the CD8+ T cells12. In arteries with CAV, the present T cells were in an activated state and expressed HLA-DR and were located in the adventitia and the neointima. Most of the CD4+ T cells were Th1, which correlates with the cytokines and chemokines that were found in the CAV arteries. Of these cytokines and chemokines TGF-β, IFN-γ and CCR4, which are known to be produced by Th1 cells, were mainly present 12, 31. 8 The study from van Loosdregt et al. shows that T cells are of significant importance in CAV. As described above, the CD4+ and CD8+ T cells are generally activated by recognition of foreign antigens. This recognition is either directly or indirectly via APCs. After activation of the CD4+/CD8+ cells, they secrete different cytokines and chemokines. Also the APCs secrete cytokines that can modulate the host T cell response to graft vascular cells11. All secreted cytokines, either secreted by APCs or by CD4+/CD8+ T cells, influence the differentiation of naïve T cells. IFN-γ for example can lead to Th1 and cytotoxic T cell (CTL) differentiation. Next to IFN-γ, transforming growth factor β (TGF-β) is present abundantly. TGF-β was found in all vascular layers31, and is a pro-inflammatory but also regulatory cytokine. TGF-β stimulates the SMCs to migrate into the intima which leads to a profibrotic state 31. Besides IFN-γ and TGF-β, IL-12 is a key cytokine. IL-12 promotes CTL differentiation from resting CD8+ T cells, but also supports Th1 initiation and NK cell activation11. Sometime after HTx, the amount of direct alloreactive T cells decreases but because the processing of foreign antigens by APCs is still proceeding, the amount of indirect alloreactive T cells increases2. From the direct alloreactive T cells, only the memory T cells remain. In CAV, many infiltrating lymphocytes express the markers of memory T cells12, 31. Memory T cells Memory T cells are believed to be important in the process of chronic allograft rejection. If recipients are primed, the memory T cells are alloreactive and cause rejection episodes32. Though it was found that memory T cells are present in CAV lesions, the exact mechanism of how memory T cells contribute to the pathogenesis of CAV is not elucidated yet. The OX40-OX40L interaction has been suggested as one of the activating pathways33. OX40-OX40L is an interaction between OX40, which is present on T cells, NK cells and neutrophils, and its ligand OX40L which is present on DCs, B cells and endothelial cells. The OX40-OX40L interaction is important for the activation of T cells in general, but also for the survival and homeostasis of memory T cells. To test if memory T cells could induce CAV, the OX40-OX40L interaction was blocked in a mouse model. After blockage, the memory T cell population in the arteries of HTx mice declined. Also the infiltration of CD4+ and CD8+ T cells in the vessel wall was reduced. This indicates that the memory T cells recruit the effector T cells33. As a consequence, these mice developed less severe CAV. Another reason why memory T cells could contribute to the development of CAV is the production of cytokines. Literature states that memory T cells secrete IFN-γ and TNF-α33. These cytokines are proinflammatory and stimulate the development of CAV. Th17 cells As shown by Hagemeijer et al., TGF-β is abundantly present in CAV lesions. TGF-β is, together with IL6 or IL-21, a differentiator of naïve T cells to Th17 cells. Furthermore, IL-6 and IL-21 are activators of signal transducer and activator of transcription 3 (STAT3). STAT3 in turn activates the retinoid-related orphan receptor γt (RORγt) or RAR-related orphan receptor C (RORC, the human equivalent of RORγt), which is a transcription factor for Th17 cells34. Th17 cells are differentiated naïve T cells which mainly produce IL-17. Already before the discovery of the Th17 cells in 2000, studies showed a correlation between IL-17 and CAV35. IL-17 is a proinflammatory cytokine and has several functions; it stimulates production of IL-6, upregulates chemoattractants, recruits neutrophils to the site of inflammation and it is also known as a profibrotic cytokine35, 36. To investigate if Th17 cells and IL-17 play a role in CAV, human endothelial cells were cultured with IL-17. Several adhesion molecules 9 increase and the inflammatory gene expression in SMCs was elevated37. This suggests that IL-17 can play a role in the development of CAV. To confirm this, Rao et al. also tested IL-17 in a model for human artery allograft rejection. In this model, immunodeficient mice underwent a transplant with human vessels. By adoptive transfer of human peripheral blood mononuclear cells (PBMCs), the mice develop an immune reaction that is similar to humans. When IL-17 was neutralized, the graft had a prolonged survival although there was still an inflammatory reaction seen. But if IL-17 was inhibited, the proinflammatory gene expression of the graft diminished significantly37. These results indicate that IL-17 contributes to CAV. However, this does not provide evidence for the involvement of Th17 cells. Recently, new results were found supporting the hypothesis that Th17 cells are included in the development of CAV. Shi and colleagues suppressed Th17 differentiation and functionality in a T-bet-/- mouse model (mice without Th1 cells received a mouse HTx). The severity of CAV was abrogated in these treated mice. The numbers of Th17 cells but also neutrophils showed no difference38. T-bet-/- mice, which did not have suppression of Th17 cells, displayed a destructive inflammatory response leading to severe CAV and huge neutrophil infiltration35. Important is to understand why the Th17 response is not inhibited by regulatory T cells (Tregs). In a healthy individual, TGF-β expression which induces Th17 cells also initiates the development and differentiation of Tregs36. Regulatory T cells Tregs are a population of T cells that prevent immune reactions against self-antigens30. The best described Treg is a CD4+ T cell that expresses CD25 and FoxP3. FoxP3 can be induced by TGF-β. But why do the Tregs not inhibit the inflammatory response that initiates CAV? One of the recently found theories is that differentiated T cells can be modified into another lineage. This means that these T cells can change in case they encounter other signals from the microenvironment. This theory was founded on the observation that Tregs became converted towards Th17 cells by IL-635. The reason for this is that before a naïve T cells becomes a fully maturated Treg, it reaches an intermediate state in which the Treg and Th17 receptors are simultaneously expressed. By addition of IL-6, the intermediate matured Treg can differentiate into a Th17 cell. As a result, the regulatory activity of the Tregs is decreased, which has implications for the Th17 response and the possible subsequent CAV development35. Research about the susceptibility of Th17 cells to Tregs demonstrated that Th17 cells are relatively resistant to the suppressive activity of polyclonal Tregs. Whereas Th1 and Th2 cells were inhibited when adding polyclonal Tregs, the Th17 cells were not suppressed by the same amount of Tregs34. However, when antigen specific Tregs were added, the Th17 cell responses were decreased. That Tregs are important in inducing tolerance is demonstrated by Ge et al. Recipient mice which were tolerant for heart allografts showed high levels of Tregs within the transplanted heart. When the Tregs were specifically deleted (specificity was based on CD25+FoxP3+ cells), the tolerance was abrogated. This abrogated tolerance might be due to the OX40-OX40L binding. OX40 costimulation between DCs and T cells inhibits TGF-β mediated Treg induction. Also if Tregs are mature, and OX40 on their membrane is occupied, the regulatory function is revoked32. Furthermore, they found that memory T cells might affect Tregs. By adding memory T cells in vitro to cultures containing naïve T cells and donor DCs, the induction of Tregs was inhibited. Blocking the expansion of memory T cells with α-OX40 antibody resulted in enhancement of Tregs and tolerant DCs. In a mouse model, Ge and 10 colleagues demonstrated that the conversion of naïve T cells to Tregs was inhibited when adding OX40 stimulated memory T cells. In conclusion, not only cytokines can affect multiple cells, also cells influence each other’s functioning. To establish if Tregs are also able to inhibit the development of CAV in humans, studies identified the ratio between effector cells and Tregs39, but also conducted experiments with human Tregs. In a study by Nadig et al., ex vivo expanded Tregs were relocated into human arteries. These human arteries were transplanted in a chimeric humanized mouse. In the human arteries which received Tregs, the in vivo development of CAV was prevented. CAV development was mainly prevented by impairing effector function and graft infiltration of leukocytes40. That Tregs inhibit effector T cells is confirmed by Roldan et al., who show that when the amount of Tregs increases, the Th1 cells decrease39. This indicates that Tregs also play a role in the human system, although more research about the human situation is required. γδ T cells are T cells which contain a T cell receptor with a γ- and a δ- chain. Most T cells have a T cell receptor which is composed of a α- and β-chain. γδ T cells make up 5% of the T cell population41. They have the ability to become active without antigen presentation, which makes them contributors to the first line of defense in tissue. Recent reports showed that host γδ T cells might accelerate both acute and chronic allograft rejection41. To investigate the role of γδ T cells in CAV, Zhu et al. used γδ T cell deficient mice which received heterotopic HTx. The γδ T cells deficiency led to a reduced infiltration of leukocytes and a decreased expression of inflammation initiators IFN-γ and HMGB1, which resulted in an increased allograft survival. Furthermore, they found that the amount of CD25+FoxP3+ Tregs was enlarged compared to the control mice. When the Tregs were depleted, the prolonged allograft survival was abrogated and the vessels in the cardiac allografts contained more severe CAV41. These results provide further evidence that Tregs play a significant role in maintaining tolerance, and when the Tregs are depleted or suppressed this leads to accelerated CAV. B cells Next to the several types of T cells, B cells are also a major contributor to adaptive immunity. B cells are mostly known about their ability to differentiate into plasma cells and produce antibodies, which also contribute to the pathogenesis of CAV. However, in 30-50% of all HTx patients with CAV, no detectable circulating antibodies are found in the transplanted heart42. This does not mean that B cells do not play a role in these patients. B cells have other functions in addition to becoming plasma cells and producing antibodies. B cells affect T cell responses by for example co-stimulation and cytokine production30. To find out if B cells might play a role in human HTx, Wehner et al. studied 16 cardiac transplants with advanced chronic rejection. They found infiltrates of B cells and plasma cells mainly in the fibrotic areas of the adventitia and the neo-intima. The infiltrates of cells were significantly more present in arteries affected by CAV than in control arteries43. Furthermore, they also found that B- and plasma cells in the adventitia formed nodules. The function of these nodules was not elucidated yet. To study the role of antibodies and B cells in CAV, Zeng and colleagues created two different mice: one mouse without antibodies and another mouse which was deficient for both antibodies and B cells. The mice which completely lacked antibodies developed CAV as compared to WT mice. But the mice which were deficient for B cells and antibodies showed minimal CAV. Furthermore, CAV was restored in mice with the B cell and antibody deficiency upon an adoptive transfer of B cells that did 11 not differentiate into plasma cells and produce antibodies42. These results suggest that B cells contribute to CAV independent of antibodies. The findings by Zeng et al. are contradictory to the results of Gareau et al. They found that antibodies produced by plasma cells are responsible for damaging SMCs in the media of coronary arteries. These results were obtained from a RAG1-/- mouse model. When the mice received antibodies via passive transfer, their arteries showed actin positive lesions. But if no antibodies were given, the B cell deficient mice did not differ significantly from the controls44. These results indicate that antibodies are important. Further investigation about the possible supporting role of B cells to T cells was performed by Zeng et al. The alloreactive T cell responses were declined in the absence of the B cells: both the T cell activity and their cytokine production were reduced. Additionally, Zeng et al. demonstrated that B cells support T cell function via antigen presentation and maintenance of splenic lymphoid architecture. The spleen is the major source of lymphocytes, if the splenic lymphoid architecture is disrupted, alloreactive T cell responses can be weakened in the transplanted heart. B cells revealed to preserve the splenic architecture and can take up alloantigen and present this to T cells. This way, B cells can contribute to the pathogenesis of CAV in an antibody independent manner. Innate immunity Next to cells from the adaptive immune system, also the innate immune system contributes to the immunologic responses that may lead to CAV. Toll like receptors CAV is correlated with persistent inflammation. Accumulating evidence states that the innate toll like receptors (TLR) contribute to the inflammatory state17. TLRs are transmembrane proteins that are expressed on dendritic cells, T- and B lymphocytes and macrophages but also on epithelial cells. They can be activated by endogenous and exogenous ligands. Which effect the activation of the TLR has is specific for the type of cell on which the TLR is present. TLRs on epithelial cells are responsible for upregulation of chemoattractants while TLRs on dendritic cells are important in naïve T cell priming22. If there is tissue damage after HTx, endogenous ligands are released. Also damage-associated molecular pattern molecules (DAMPs) such as heat shock protein 70 (HSP70) and high-mobility group box 1 (HMGB1) are produced45. These endogenous ligands signal via TLRs, mostly TLR4. This way, the molecules that are secreted after tissue or ischemia-reperfusion injury can promote a TLR4dependent response or stimulate proinflammatory molecules such as TNF and IL-6. This excites the general alloimmune response. Via these mechanisms, the TLRs can be activated after HTx and stimulate both the innate and the adaptive immune response, which can lead to rejection. Macrophages Macrophages are professional antigen presenting cells that act both in the innate and in the adaptive immune response. The exact role of the macrophages in CAV is not known. However, it is plausible that macrophages play a role in the initiation of the adaptive immune response since macrophages secrete HMGB1 in response to cytokines, especially IFN-γ and TNF, and TLR ligands11. HMGB1 can act on human T cells by binding to its receptor Receptor for Advanced Glycation Endproducts (RAGE), which is present on T cells. Binding of HMGB1 influences cytokine production or T cell differentiation11. Macrophages also contain Fc receptors, which are capable to respond to antibodies 12 that recognize foreign DSA or complement factors. Besides this, macrophages are also competent to produce growth factors and chemokines, which possibly stimulates the proliferation of the neointima46. To establish the potential role of macrophages, Kitchens et al. depleted or defunctionalized (by inhibition of phagocytosis) macrophages in mice after HTx. Depletion of macrophages reduced the development of CAV, while inhibition of the phagocytotic capabilities of the macrophages failed to abrogate the process. Also delayed depletion of the macrophages did not diminish CAV. This indicates that the macrophages are significant in the initiation of CAV, while they seem not to be required in the maintenance of the CAV lesions46. Another possible role of macrophages is due to their ability to produce matrix metalloproteinases (MMPs) 47. Among the produced MMPs are MMP-2 and MMP-9, these MMPs are involved in the remodeling and activation of fibrosis. Huibers et al. showed that MMP-2 and MMP-9 were expressed in CAV arteries. This indicates that the MMP production of macrophages might contribute to the pathogenesis of CAV47. Natural killer cells An important player in innate immunity is the natural killer (NK) cell. The NK cell is a lymphocyte that is able to recognize foreign cells without priming or prior exposure to the particular antigen. The activation of a NK cell is regulated by inhibitory and activating receptors that are expressed on the membrane of the NK cell. The inhibitory receptor on the NK cell recognizes non-self cells by the absence of a self-MHC molecule on the surface. The activating receptors recognize cell surface molecules that are induced by viral infection or DNA damage30. If the balance is leaning to activation, the NK cell kills the target cell by the release of perforin or granzymes. The NK cell simultaneously initiates the innate immune response by secreting cytokines as IFN-γ. Until a few years ago, the general concept was that NK cells do not contribute to rejection of solid organs. For example, the study from Hagemeijer et al. shows that there are no NK cells found in arterial walls with CAV. In this study, they characterize the infiltrating mononuclear cells in the human arterial walls. This infiltrating population consisted of various different cell types such as Bcells, macrophages, CD4+ and CD8+ T cells. However, no NK cells were found in the CAV arteries or in the extra vascular mononuclear cell population31. Although Hagemeijer et al. did not find NK cells in the human arterial walls, another study did find infiltration of NK cells. In 2002 Ankersmit et al. compared twenty patients with CAV with twenty HTx patients without CAV. They saw a significant increase in NK cells in the allograft of CAV patients compared to control48. Despite these conflicting results, there is accumulating evidence that NK cells are involved in the development of CAV after HTx. Hirohashi and colleagues elicited CAV by adoptive transfer of donor specific antigen (DSA) to MHC-I molecules in mice which do not have mature T- and B lymphocytes (RAG-/-). They discovered that after depletion of NK cells, the mice did not develop CAV. If solely the mature NK cells were depleted, the severity of CAV decreased significantly. When analyzing the CAV lesions, they found that these were infiltrated by NK cells and macrophages49. Furthermore, a short experiment with parental-to-F1 HTx demonstrated that NK cells are involved in the development of CAV50. With the parental-to-F1 HTx, the offspring of BALB/c and C57BL/6 mice, F1 hybrid mice, received a heart from the C57BL/6 mice. Even though the F1 mice are tolerant for the donor antigens, because the allograft is derived from one of the parents, CAV was prospering. As a control, heart isotransplants were conducted. These mice received the heart of genetically identical individual. In none of the heart isotransplants was CAV found, while in the F1 hybrid mice 19 of the 13 22 hearts showed advanced CAV50. By staining with a NK cell specific antibody, also the presence of the NK cells in the lesions was established. To find out if these NK cells could be responsible for inflammation and with that for CAV, the levels of NK cell cytotoxicity were compared between the isotransplants and the F1 hybrid mouse transplants. The cytotoxicity of the NK cells in the F1 hybrid mice was significantly higher compared to control50. Recipients which were IFN-γ deficient did not develop CAV. These data support the hypothesis that NK cells contribute to the pathogenesis of CAV, but the presence of IFN-γ is mandatory. The finding that IFN-γ is required for the development of CAV indicates that NK cells can promote an antigen specific CD8+ T cell response by supporting the maturation of dendritic cells or by direct action of secreted cytokines such as IFN-γ45. This discovery represents a link between the innate and the adaptive immune system. NK cells also have been implicated in the pathogenesis of virus-induced CAV. Research performed by Gramham and colleagues suggests a NK cell dependent development of CAV after inoculation of a specific virus (lymphocytic choriomeningitis virus: LCMV). LCMV is correlated with increased NK cell activity and establishes a non-lethal infection in mice. If patients get LCMV after HTx, graft tolerance is reduced18. By using the parent-to-F1 strategy in combination with LCMV inoculation in Rag-/- mice, the role of NK cells and the status of CAV in the arteries was measured. Twenty-eight days after HTx, both NK cells and macrophages were found in the CAV lesions of the mice. To test if the NK cells are essential for the progression of CAV, the NK cells were depleted with a NK1.1 antibody. Fifty-six days after HTx, NK cell depletion was higher than 80%. From these mice, which were infected with LCMV and treated with the NK1.1 antibody, only 1 out of 7 developed CAV18. Overall, these studies suggest that NK cells do play a significant role in the pathogenesis of CAV. However, these functional studies about NK cells are only performed in mice. No human studies about the functional mechanism of NK cells in CAV have been accomplished. Complement system The complement system is a mechanism that is composed by three pathways: the classical, the alternative and the lectin pathway30. It can amplify the reaction of both the innate as the adaptive immune system. The complement system is in the pathogenesis of CAV mainly activated by the deposition of DSA to the graft endothelial cells28. Human vascular endothelial cells and SMC are sensitive to various components of the complement system. This can influence the differentiation and proliferation of the intima, which can finally lead to fibrosis51. The complement system can modulate a variety of cells among which macrophages, T- and B cells thereby contributing to the process of CAV51. 14 Figure 2. Immunological mechanisms in CAV Overview of the most important immune cells involved in the development of CAV. The transplanted heart (allograft) is recognized by antigen presenting cells (APC), macrophages and natural killer cells (NK cell). The foreign antigen is taken up by APCs and presented to CD8+ and CD4+ T cells. Macrophages and NK cells take up antigen en attack the allograft directly and/or stimulate CD8+ T cells. Produced cytokines induce regulatory T cells (Treg) and memory T cells (Tmem). The Treg inhibits the activation of Th17 and Tmem cells. B cells do not only differentiate into plasma cells to make antibodies but also support CD8+ T cells. Th17, Tmem, CD8+ T cells together with antibodies can lead to rejection of the transplanted heart. 15 Chapter 3: Fibrosis Activation of all these immunological mechanisms has great impact on the heart allograft. Cytokines and chemokines, but also other pro- and anti-fibrotic factors are secreted by immune cells and influence the various layers of the arterial wall. Immunological mechanisms in CAV can lead to coronary artery fibrosis. As shortly mentioned before, CAV is characterized by intimal thickening due to accumulation of smooth muscle cells (SMCs) and additional extracellular matrix (ECM). The internal and external elastic laminae stay intact47, 52. In early stages of CAV but mostly in later stages, vessels which are affected by CAV show outward compensation. The vessel is remodeling to preserve the lumen where the intima is thickening (Huibers et al., 2014 accepted Atherosclerosis). This can correlate with reduced endothelial function and SMC contractility11. When the intimal thickening can no longer be compensated, the remodeling stops. Although there has been many studies about CAV, there is some discussion about the pathology of arteries with CAV. Some research shows that the neo-intima is composed out of two layers: a luminal layer (NI-LL), which is composed out of loose connective tissue with infiltrated mononuclear cells, and a SMC layer (NI-SMC) which contains SMCs or myofibroblasts12, 47, 53. While others state that the neo-intima exists in just one layer 52. Despite these differences, the general concept is that the intima is thickened. To study CAV, the differences between animals and humans must be considered. In mice which underwent HTx, the neo-intima is more rapidly formed than in humans. Furthermore, this neo-intima contains mainly SMCs54, while in humans it also contains connective tissue filled with mononuclear cells12. Endothelial cells and fibrosis The endothelial layer in the coronary vessel wall plays an essential role in the perception of signaling and changes in blood pressure. As a response, the endothelium releases vasoactive molecules that regulate vascular relaxing or contracting to maintain vascular homeostasis13. When homeostasis is unbalanced, the endothelial layer is more prone to processes as disturbed coagulation, adhesion of leukocytes and vascular inflammation. In patients who underwent HTx, the endothelium of the donor heart is the first biological compound the host immune system will recognize. The immune response of the host determines the gradient of rejection of the allograft. There are a few theories about the role of the endothelium in this process. One of the theories describes how endothelial cells of the donor are replaced by endothelial cells of the host in response to tissue injury. Hollenberg et al. showed in a study with seventy-three patients that endothelial dysfunction in the coronary arteries predicted CAV after HTx55. They illustrated that the local availability of nitric oxide (NO) was reduced in patients with endothelial dysfunction. Normally, NO inhibits platelet and leukocyte adherence to the endothelial wall by suppressing adhesion molecules and chemokines. Reduction of NO initiated and progressed the potential for blood platelets and leukocytes to adhere, and for platelet-derived growth factor (PDGF) to increase in concentration55. Adherence of platelets and leukocytes leads to local oxidative stress and the stimulation of inflammatory genes which respond to oxidants. Furthermore, NO plays a significant role in the proliferation of SMCs. If NO is decreased, the SMCs proliferate more and the neo-intima thickens. By the secretion of inflammatory cytokines by leukocytes, it is hypothesized that the SMCs are activated and migrate from the media to the intima (donor derived) or from the circulation to the intima (host derived) 13, 53. Probably, there are also interactions between the endothelial layer of the donor and the endothelium of the host. The precise mechanism of these interactions is however not fully elucidated 16 yet. A few studies indicate that endothelial cells from the host integrate in the endothelium of the graft55, 56. These host endothelial cells then partly replace the graft endothelial layer. If the graft endothelial layer is damaged, the endothelial cells are discarded, they might undergo apoptosis and circulate in the blood. The progenitor cells then migrate to the site of injury and promote the repair process. This is partly mediated by NO55. To trace the progenitor cells from the host, males who underwent an HTx with female hearts were investigated. By in situ hybridization with the Ychromosome, the host cells could be followed into the female heart56. In the study of Minami et al. the donor endothelial cells were for 24% replaced by recipient cells. They also found, coherent with the results of De Weger et al., that the endothelial cell replacement was mostly present in the small epicardial and intramyocardial vessels, the same vessels as where CAV is generally allocated56, 57 The role of smooth muscle cells in fibrosis The majority of cells that are present in a thickened intima are SMCs. These SMCs are present in the NI-SMC layer47. The increased amount of SMCs in the thickened intima might have different sources: the intima, the SMCs are simply proliferated; the SMCs are migrated from the media to the intima, this is mostly shown in animal studies23, 53; the SMCs are derived from progenitor cells present in the medial/adventitial border or the SMCs are derived from recruited circulating host cells3. Because there are several theories about the source of the SMCs in the neo-intima, studies have tried to identify the surface markers of these SMCs. By comparing the surface markers of the SMCs in the neo-intima and the media, a possible origin of the neo-intimal SMCs can be identified. In these studies, it was observed that neo-intimal SMCs showed a different phenotype than medial SMCs, mostly the chemokine receptor expression differed58. To study the exact source of the neo-intimal cells, human coronary arteries from CAV patients were histologically analyzed. The results from this study indicated that the neo-intimal SMCs are derived from pre-existing donor SMCs or myofibroblasts54. Circulating progenitor cells were not involved in the formation of neo-intimal SMCs in this patient group. This is contradictory to several animal studies, in which was found that host progenitor cells do play a role in neo-intimal SMC formation54, 58 . In animals, the media is often thinning by necrosis. This process is rarely seen in humans. Therefore, the media cannot be a large source of SMCs in animal studies. This is also demonstrated in a study by Zheng et al. were they show that progenitor cells are recruited from circulation by several chemokines. These host progenitor cells proliferate and differentiate in neo-intimal SMCs58. Most likely, the media is also preserved in humans by the immunosuppressive therapy that is given after HTx. These medial SMCs can be a source for intimal thickening. Another study by Devitt et al. demonstrates a role for the so called process of ‘benign intimal thickening’ (BIT). In this new theory, the normal human coronary arteries contain a thickened intima due to SMCs. This layer developed during life and is lacking in animals. By ischemia-reperfusion injury after transplantation, this layer is damaged which causes activation of the immune system with as result CAV53. The BIT layer is also present in the transplanted heart. Devitt and colleagues show that the donor BIT layer is preserved for several years, which makes it a possible source of intimal cells53. All these studies suggest sources for the neo-intimal SMCs. But what are the triggers that cause neo-intima thickening and why do the SMCs proliferate? 17 AIF-1 Data from several studies indicate a role for allograft inflammatory factor-1 (AIF-1). AIF-1 is a cytoplasmic calcium-binding protein which is expressed in medial and neo-intimal SMCs in allograftinjured arteries59. AIF-1 is solely produced in response to inflammatory cytokines such as IFN-γ and TGF-β60. The expression of AIF-1 was correlated with the development of CAV, which was measured in rejected hearts. Furthermore, a significant correlation was found between the amount of AIF-1 and intimal thickening60. AIF-1 stimulates intimal thickening by triggering SMCs to migrate. This is established by its binding to actin and activation of RAC1 (Ras-related C3 botulinum toxin substrate 1). RAC1 is a protein that stimulates motility of the cell via actin. Upon binding of AIF-1 actin polymerizes. The combination of polymerization of actin and RAC1 activation causes the SMCs to migrate59. Next to migration, AIF-1 also stimulates SMC proliferation by inducing cytokine granulocyte-colony stimulating factor (G-CSF). G-CSF showed to induce human SMC proliferation in vitro. To study the link between G-SCF and AIF-1, cells were seeded which contained AIF-1 or no AIF1. Addition of a G-SCF neutralizing antibody inhibited the proliferation of SMCs in cells containing AIF-1. In cells without AIF-1, the neutralizing antibody did not give any effects59. This shows that GSCF has effects on AIF-1. Next to AIF-1, growth factors such as platelet-derived growth factor (PDGF) and transforming growth factor (TGF) stimulate SMC proliferation and vascular remodeling. PDGF not only has a strong chemotactic and mitogenic effect on SMCs but also on fibroblasts61, 62. It was shown to be produced by cells that are involved in the inflammatory process and endothelial injury. The expression of PDGF was significantly increased in arteries that were affected by CAV61. Furthermore, the amount of collagenous fibers in the coronary arteries correlated with the severity of rejection61. By inhibiting PDGF with an antibody, the development of CAV was inhibited. This suggests that PDGF plays a role in CAV and fibrosis. Recent research by Iida et al. demonstrates that PDGF stimulates SMC proliferation and migration in vitro via activation of Adenosine monophosphate-activated protein kinase (AMPK). AMPK regulates several pathways among which the MAP-kinase pathway and the PI3-kinase/Akt pathway. These pathways lead to cell proliferation, differentiation, survival and migration63. Although PDGF is expressed in low levels in the wall of healthy arteries, its production is increased after endothelial injury as a result of HTx63, 64. All cells that make up the normal arterial wall and infiltrating immune cells can produce PDGF64. The increased production of PDGF after HTx also indicates that PDGF could play a role in SMC proliferation and migration leading to intimal thickening. Other research demonstrates that all PGDF ligands and receptors are upregulated in the intima during chronic rejection, which resulted in increased TGF-β production in fibroblasts62. Furthermore, a PDGF gene transfer in a rat CAV model also led to a two-fold increase in cardiac fibrosis. By upregulation of PDGF ligands on SMCs, induced by among others TGF-β, the SMCs are more susceptible for the mitogenic signals of PDGF62. Immune cells causing fibrosis Next to endothelial cells and SMCs, also immune cells contribute to fibrosis. Immune cells mainly contribute to fibrosis by secretion of cytokines and chemokines. 18 TGF-β and related cytokines Huibers et al. showed that in CAV, fibrosis mainly takes place in the NI-LL47. They found only a small difference in expression of fibrotic factors between CAV en controls. However, they did find a specific disparity in the localization of TGF-β. TGF-β was not only present in stromal cells but also secreted by active T cells that infiltrated the intima. This provided larger amounts of TGF-β in the CAV vessels47. Other research demonstrates that TGF-β promotes fibrosis via T cell signaling36. To study the role of TGF-β, the CD4+ T cells were depleted in a vascular mouse model. As the T cells (Th1, Th2, Th17 and CD8+) infiltrated the allograft and responded to TGF-β, fibrosis was promoted. When the effector T cells were made unresponsive to TGF-β, the fibrosis in the grafts was minimal36. This indicates that TGF-β contributes to the fibrotic process but is not sufficient to initiate the process without other immune cells. Furthermore, TGF-β upregulates the collagen synthesis in SMCs. Collagen is one of the most abundant molecules present in the thickened intima65. Another contribution to the fibrotic response is possibly due to fibrosis stimulating factors like PDGF, as mentioned in the previous paragraph, but also by locally increasing MMPs. MMPs in their turn create a positive feedback loop for intimal thickening by boosting the availability of TGF-β65. Next to the direct effects of TGF-β, it also induces modulator proteins that enhance its effects. One of these proteins is connective tissue growth factor (CTGF)65. CTGF is secreted in SMCs in the ECM and is found to be an important mediator in the development of interstitial fibrosis. CTGF is mainly found in the NI-LL and the adventitia of human coronary arteries with CAV47. TGF-β activates the transcription factor SMAD3 (small mother against decapentaplegic 3) which activates the transcription of CTGF. After secretion of CTGF, it increases ECM production, fibroblast proliferation, and enhancement of adhesion by other cells such as leukocytes. Thus, activation of CTGF results in an increased fibrotic area66. TGF-β is part of the superfamily of TGF-β. Another subfamily of the TGF-β superfamily is bone morphogenic protein (BMP). BMP can also activate transcription factors of the SMAD group. Since BMP binds with different affinity to SMAD proteins, the activation gives a different outcome then when TGF-β activates SMAD proteins67. Also cell type and environmental triggers affecting the cell influence the actions of SMAD proteins. As binding of TGF-β to SMAD promotes fibrosis in the heart, binding of BMP to SMAD provides beneficial effects for the heart67. For instance, BMP-2 has antiapoptotic features whereas BMP-4 and BMP-7 are correlated with anti-fibrotic effects47, 67. Research by Zeisberg et al. shows that BMP-7 preserves the endothelial phenotype and reverses the actions of TGF-β. According to several researchers, endothelial cells undergo an endothelial-mesenchymal transition (EndMT) which means that endothelial cells differentiate into cardiac SMC-like and fibroblast-like cells68, 69. This transition is initiated under influence of TGF-β. After addition of BMP-7 in a mouse model, the endothelial phenotype was preserved68. Mice with an impaired SMAD3 signaling pathway showed a decrease in fibrosis, indicating that TGF-β stimulates fibrosis68. These results indicate that the severity of fibrosis is determined by the balance between TGF-β and BMP-7 and the preservation of endothelial function. Recently it was found that suppressor of cytokine signaling 1 (SOCS1) is also a preserver of endothelial function70. SOCS1 is induced as a response to proinflammatory cytokines that are released by infiltrating immune cells. The activation of SOCS1 results in a decrease in cytokine production. SOCS1 expression by endothelial cells showed to be essential for the preservation of 19 endothelial function. When SOCS1 was absent in a mouse aorta transplantation model, the endothelial cells showed a reduced relaxation in response to addition of a vasodilator. Additionally, the constriction of the endothelial cells increased as a reaction to addition of a vasoconstrictor70. This indicates that SOCS1 is important for the functioning of endothelial cells. IFN-γ One of the most important proinflammatory cytokines that is produced during inflammation is IFN-γ. IFN-γ is produced by several cell types such as CD8+ T cells, Th1 cells and NK cells. Convincing evidence that IFN-γ is involved in CAV is provided by van Loosdregt et al. They showed that not only IFN-γ but also IFN-γ inducible chemokines, such as RANTES (regulated on activation, normal T cell expressed and secreted) and ITAC (Interferon-inducible T-cell alpha chemoattractant), were present in the arteries containing CAV12. Furthermore, IFN-γ and its chemokines were found in the intima and the adventitia but not in the media. IFN-γ has effects on various cells including endothelial cells and SMCs to produce chemokines. Although SMCs are activated by IFN-γ in the same way as endothelial cells, they do not activate T cells and can inhibit T cells that respond to the endothelial cells11. Because the SMCs are present in great numbers in the media, this might be the reason why the media layer of the vessel wall is the least infiltrated by T cells in CAV. Via the induction of chemokines, IFN-γ can modulate the immune response. Some of these chemokines bind to CXCR3 receptors which in turn attract potent antigen-primed CD8+ T cells and macrophages71. Next to cells of the immune system, IFN-γ inducible chemokines can also modulate vascular cells. By way of several mechanisms, which are to detailed to discuss in this review, the IFN-γ induced chemokines stimulate angiogenesis and growth factor-generated SMC proliferation71. Both of the effects of IFN-γ inducible chemokines add to the development of CAV. Furthermore, it was found in a human artery graft mouse model that IFN-γ is also able to induce cell proliferation and cell death of SMCs11. IFN-γ primes SMCs to Fas-induced apoptosis by relocation of Fas to the cell surface. Apoptosis Apoptosis can also lead to fibrosis. Apoptotic cells have been identified as significant players in the initiation of fibrosis. In fibrosis during CAV, SMCs and endothelial cells that undergo apoptosis are the main initiators of fibrosis. Due to the immune responses that are present during rejection, SMCs and endothelial cells of the donor heart are eradicated. The eradication can be completed via the intrinsic or the extrinsic apoptosis pathways72. The intrinsic pathway is induced by for example perforin, which is secreted by NK cells. Due to perforin, an intracellular apoptotic cascade is activated. The extrinsic pathway works via Fas-Fas ligand interaction and can be influenced by TGF-β. Apoptotic cells can either directly or indirectly stimulate fibrosis. Directly by paracrine signaling and indirectly by stimulating immune cells to secrete factors that induce fibrosis73. Extra immune cells are attracted by upregulation of adhesion molecules on the endothelium. Apoptotic endothelial cells signal to neighboring cells, which as a result upregulate ICAM-1 and VCAM-172. An aspect of direct stimulation of fibrosis via apoptotic cells is induction of ECM proteolysis. Endothelial cells that are in the first stages of apoptosis, adjust their surface proteoglycans thereby initiating ECM proteolysis72. As a result, the endothelial cells can detach and end up in the circulation. 20 Proteolysis of the ECM leads to the release of certain molecules that are aimed at preventing neighboring cells to go into apoptosis. These factors however can also lead to increased survival of fibroblasts and myofibroblasts, enhancing the fibrotic process72. The immune system can also be modulated by apoptotic cells via phagocytosis of the apoptotic cells by macrophages. After phagocytosis of apoptotic cells, the macrophages can change from phenotype. As a result of this phenotype-switch, the macrophages becomes less inflammatory and secrete factors that stimulate tissue growth. For example, TNF-α and IL-6 are lowered while IL-10 and TGF-β are upregulated 73. IL-10 and TGF-β are known to stimulate cell survival in the surrounding tissue. With this, these cytokines could trigger a fibrotic response by stimulating apoptosis resistance and support fibroblasts and myofibroblasts to proliferate and differentiate73. Figure 3. Fibrotic mechanisms in CAV After heart transplantation, endothelial damage is likely to occur. As a result, endothelial cells loose function. Adhesion molecules are upregulated and leukocytes transmigrate through the endothelial layer. There they produce several cytokines among which IFN-γ or phagocytose apoptotic endothelial cells. After phagocytosis, TGF-β is produced. TGF-β stimulates several processes: production of AIF-1 and PDGF, activation of SMAD proteins, resistance to apoptosis and upregulation of collagen production by SMCs. The SMAD proteins that are activated by TGF-β stimulate CTGF production by SMCs, which causes increased proliferation and migration of SMCs. SMAD proteins that are activated by BMP inhibit these SMC processes. The increased collagen production, resistance to apoptosis and increased proliferation and migration of SMCs lead to fibrosis. 21 Discussion In summary, CAV is a disease which has a high prevalence in HTx patients. It is characterized by concentric thickening of the coronary artery wall of the transplanted heart. The pathogenesis of CAV is complex; various cell types from both the host and the donor play a role. CAV can be initiated by several causes among which are I-R injury and CMV infection. These causes or risk factors trigger the host’s immune system. The innate and the adaptive immune system are activated and cooperate to attack the foreign allograft. Macrophages and NK cells recognize the HLA molecule of the transplanted heart via TLRs as foreign and attack it. APCs identify antigens on the transplanted heart, take them up and present them to CD8+ and CD4+ T cells. Next to these cells, cytokine production stimulates regulatory, Th17 and memory T cells to be activated. Tregs are able to induce tolerance by inhibiting the immune response via CD8+ T and Th17 cells. If memory T cells are formed they can recruit CD8+ T cells. Besides T cells and innate immune cells, B cells also contribute to CAV. B cells add to the pathogenesis of CAV by supporting T cell function but also by differentiating into plasma cells that produce antibodies. These antibodies are able to damage SMCs and with that initiate CAV. Activation of immunological components affects the vasculature of the heart allograft. Inflammation causes dysfunction of the endothelial layer which results in more adhesion of leukocytes and less availability of NO. This reduced NO contributes to the proliferation of SMCs. Furthermore, the production of cytokines and several growth factors by immune cells and endothelial cells stimulate the migration and proliferation of SMCs. The continuing SMC proliferation leads to intimal thickening. Additionally to the proliferation of SMCs, the produced growth factors and cytokines can also trigger fibrosis by stimulating fibroblast proliferation and survival. Besides these just named processes, apoptosis is also important in the development of CAV. Apoptotic endothelial cells and SMCs stimulate fibrosis via the induction of fibroblasts and myofibroblasts, but also by stimulating immune cells to produce pro-fibrotic factors. In the past few decades, a lot of knowledge is gained about the processes that underlie the pathogenesis of CAV. Despite these new insights there are still gaps in our knowledge about CAV in humans. Many studies are performed in mouse models, studies in human cardiac transplant recipients are limited. Although mouse models can be very useful, differences between mice and humans remain. CAV develops in mice already within 14 days while in humans it is a process that takes years to develop12. Furthermore, the composition of a CAV lesion and the immune response differs between animals and humans. For instance the timing and production of cytokines is different in experimental animals74. To partly solve this problem, more humanized mouse models, such as human-mouse chimeric artery mice, should be used. This way, the human immune responses to HTx in the research models are more closely related to the occurring processes in patients. Another issue that still needs to be addressed is the exact role that B cells play in the development of CAV in humans. Contradictory results are found about B cells. One study shows that B cells act mainly in an antibody-independent way42, while other research states that the primary function of B cells is to differentiate into plasma cells and secrete antibodies44. The B cell mechanism was solely studied in mice. In humans, just the presence of B cells in CAV lesions is established43. Although Wehner et al. found that the B- and plasma cells were present in nodules in the coronary arteries, the role that these infiltrates play in CAV is not known yet. There might be a functional reason for B- and plasma cells to form nodules. 22 The same problem also holds for NK cells and γδ T cells. No human studies are accomplished about the contribution of NK cells and γδ T cells to CAV in humans. To study the role of NK cells, often mice are used that lack functional T and B lymphocytes. In patients who develop CAV, all lymphocytes are of course operative. The interactions between NK cells and T and B cells need to be more elucidated to gain more insight about the role of NK cells in the pathogenesis of CAV in humans. γδ T cells have shown to contribute to tolerance of liver transplants in humans75. Therefore, the hypothesis was proposed that γδ T cells are also involved after HTx. Now that Zhu et al. have shown in a mouse model that these cells not only have direct effects on allograft arteries but also interact with Tregs, further research regarding γδ T cells is interesting. Research about γδ T cells is not just important for elucidating their role in CAV, but also to find out more about the contribution of Tregs in a human setting. To elucidate the exact mechanisms of the many processes that are involved in the development of CAV, more humanized mouse models should be used. Furthermore, the interactions between cells from the innate and the adaptive immune system have to be investigated. Within these interactions, mechanisms might be present that contribute to the initiation and progress of CAV that we are not yet familiar with. Another possibility is that immune cells that nowadays are not correlated with CAV play a role. An example is the finding a few years ago, when the γδ T cells were found to be involved in CAV. Furthermore, we have to keep in mind that the contribution of cells or cytokines to CAV is almost never black and white. The key to prevent CAV is maintaining the balance between pro- and anti-inflammatory and fibrotic factors. One example of this is TGF-β, which is not only a proinflammatory cytokine but has also been shown to contribute to an allograft-tolerant state36. Besides the immunological side of CAV, to find out the origin of the migrating SMCs is important for understanding how the intimal thickening evolves. Additionally, the factors that are secreted by SMCs but also by endothelial cells have to be researched more to establish all their functions. For therapy, we believe there is a gap in knowledge about Tregs. If we can find out how to maintain and control the regulatory capacities from Tregs, this might be the solution to limit excessive inflammation including all the consequences. 23 References 1. Taylor DO, Edwards LB, Boucek MM, et al. Registry of the International Society for Heart and Lung Transplantation: twenty-fourth official adult heart transplant report--2007. J Heart Lung Transplant. 2007;26(8):769-781. 2. Angaswamy N, Tiriveedhi V, Sarma NJ, et al. Interplay between immune responses to HLA and nonHLA self-antigens in allograft rejection. Hum Immunol. 2013;74(11):1478-1485. 3. Pober JS, Jane-Wit D, Qin L, Tellides G. Interacting Mechanisms in the Pathogenesis of Cardiac Allograft Vasculopathy. Arterioscler Thromb Vasc Biol. 2014. 4. Costello JP, Mohanakumar T, Nath DS. Mechanisms of chronic cardiac allograft rejection. Tex Heart Inst J. 2013;40(4):395-399. 5. Berry GJ, Burke MM, Andersen C, et al. The 2013 International Society for Heart and Lung Transplantation Working Formulation for the standardization of nomenclature in the pathologic diagnosis of antibody-mediated rejection in heart transplantation. J Heart Lung Transplant. 2013;32(12):1147-1162. 6. Rose ML. Role of anti-vimentin antibodies in allograft rejection. Hum Immunol. 2013;74(11):14591462. 7. Tonsho M, Michel S, Ahmed Z, Alessandrini A, Madsen JC. Heart transplantation: challenges facing the field. Cold Spring Harb Perspect Med. 2014;4(5):10.1101/cshperspect.a015636. 8. Angelini A, Castellani C, Fedrigo M, et al. Coronary cardiac allograft vasculopathy versus native atherosclerosis: difficulties in classification. Virchows Arch. 2014;464(6):627-635. 9. Alexander Eskandary F, Kohl M, Dunkler D, et al. Lack of donor and recipient age interaction in cardiac transplantation. J Heart Lung Transplant. 2014;33(6):629-635. 10. Valantine H. Cardiac allograft vasculopathy after heart transplantation: risk factors and management. J Heart Lung Transplant. 2004;23(5 Suppl):S187-93. 11. Tellides G and Pober JS. Interferon-gamma axis in graft arteriosclerosis. Circ Res. 2007;100(5):622-632. 12. van Loosdregt J, van Oosterhout MF, Bruggink AH, et al. The chemokine and chemokine receptor profile of infiltrating cells in the wall of arteries with cardiac allograft vasculopathy is indicative of a memory T-helper 1 response. Circulation. 2006;114(15):1599-1607. 13. Colvin-Adams M, Harcourt N, Duprez D. Endothelial dysfunction and cardiac allograft vasculopathy. J Cardiovasc Transl Res. 2013;6(2):263-277. 14. Sanchez Lazaro IJ, Almenar Bonet L, Moro Lopez J, et al. Influence of traditional cardiovascular risk factors in the recipient on the development of cardiac allograft vasculopathy after heart transplantation. Transplant Proc. 2008;40(9):3056-3057. 15. Vassalli G, Gallino A, Weis M, et al. Alloimmunity and nonimmunologic risk factors in cardiac allograft vasculopathy. Eur Heart J. 2003;24(13):1180-1188. 24 16. Wehner JR and Baldwin WM,3rd. Cardiac allograft vasculopathy: do adipocytes bridge alloimmune and metabolic risk factors? Curr Opin Organ Transplant. 2010;15(5):639-644. 17. Schmauss D and Weis M. Cardiac allograft vasculopathy: recent developments. Circulation. 2008;117(16):2131-2141. 18. Graham JA, Wilkinson RA, Hirohashi T, et al. Viral infection induces de novo lesions of coronary allograft vasculopathy through a natural killer cell-dependent pathway. Am J Transplant. 2009;9(11):2479-2484. 19. Streblow DN, Orloff SL, Nelson JA. Acceleration of allograft failure by cytomegalovirus. Curr Opin Immunol. 2007;19(5):577-582. 20. Weis M and Cooke JP. Cardiac allograft vasculopathy and dysregulation of the NO synthase pathway. Arterioscler Thromb Vasc Biol. 2003;23(4):567-575. 21. Potena L, Holweg CT, Chin C, et al. Acute rejection and cardiac allograft vascular disease is reduced by suppression of subclinical cytomegalovirus infection. Transplantation. 2006;82(3):398405. 22. Obhrai J and Goldstein DR. The role of toll-like receptors in solid organ transplantation. Transplantation. 2006;81(4):497-502. 23. Devitt JJ, King CL, Lee TD, Hancock Friesen CL. Early innate immune events induced by prolonged cold ischemia exacerbate allograft vasculopathy. J Cardiothorac Surg. 2011;6:2-8090-6-2. 24. Colvin-Adams M and Agnihotri A. Cardiac allograft vasculopathy: current knowledge and future direction. Clin Transplant. 2011;25(2):175-184. 25. Crudele V, Cacciatore F, Grimaldi V, et al. Human leukocyte antigen-DR mismatch is associated with increased in-hospital mortality after a heart transplant. Exp Clin Transplant. 2013;11(4):346-351. 26. Cohen O, De La Zerda DJ, Beygui R, Hekmat D, Laks H. Donor brain death mechanisms and outcomes after heart transplantation. Transplant Proc. 2007;39(10):2964-2969. 27. Eskandary FA, Kohl M, Dunkler D, et al. Lack of donor and recipient age interaction in cardiac transplantation. J Heart Lung Transplant. 2014;33(6):629-635. 28. Jane-Wit D, Manes TD, Yi T, et al. Alloantibody and complement promote T cell-mediated cardiac allograft vasculopathy through noncanonical nuclear factor-kappaB signaling in endothelial cells. Circulation. 2013;128(23):2504-2516. 29. Guihaire J, Mercier O, Flecher E, et al. Comparison of cardiac allograft vasculopathy in heart and heart-lung transplantations: A 15-year retrospective study. J Heart Lung Transplant. 2014;33(6):636643. 30. Kumar V, Abbas A, Fausto N, Aster J. Robbins and Cotran Pathologic basis of disease. ; 2010. 25 31. Hagemeijer MC, van Oosterhout MF, van Wichen DF, et al. T cells in cardiac allograft vasculopathy are skewed to memory Th-1 cells in the presence of a distinct Th-2 population. Am J Transplant. 2008;8(5):1040-1050. 32. Ge W, Jiang J, Liu W, et al. Regulatory T cells are critical to tolerance induction in presensitized mouse transplant recipients through targeting memory T cells. Am J Transplant. 2010;10(8):17601773. 33. Wang H, Zhang Z, Tian W, et al. Memory T Cells Mediate Cardiac Allograft Vasculopathy and are Inactivated by Anti-OX40L Monoclonal Antibody. Cardiovasc Drugs Ther. 2014;28(2):115-122. 34. Heidt S, Segundo DS, Chadha R, Wood KJ. The impact of Th17 cells on transplant rejection and the induction of tolerance. Curr Opin Organ Transplant. 2010;15(4):456-461. 35. Chadha R, Heidt S, Jones ND, Wood KJ. Th17: contributors to allograft rejection and a barrier to the induction of transplantation tolerance? Transplantation. 2011;91(9):939-945. 36. Faust SM, Lu G, Marini BL, et al. Role of T cell TGFbeta signaling and IL-17 in allograft acceptance and fibrosis associated with chronic rejection. J Immunol. 2009;183(11):7297-7306. 37. Rao DA, Eid RE, Qin L, et al. Interleukin (IL)-1 promotes allogeneic T cell intimal infiltration and IL17 production in a model of human artery rejection. J Exp Med. 2008;205(13):3145-3158. 38. Shi X, Zhang M, Liu F, et al. Tim-1-Fc suppresses chronic cardiac allograft rejection and vasculopathy by reducing IL-17 production. Int J Clin Exp Pathol. 2014;7(2):509-520. 39. Roldan C, Mirabet S, Brossa V, et al. Correlation of immunological markers with graft vasculopathy development in heart transplantation. Transplant Proc. 2012;44(9):2653-2656. 40. Nadig SN, Wieckiewicz J, Wu DC, et al. In vivo prevention of transplant arteriosclerosis by ex vivoexpanded human regulatory T cells. Nat Med. 2010;16(7):809-813. 41. Zhu H, Li J, Wang S, Liu K, Wang L, Huang L. gammadelta T cell receptor deficiency attenuated cardiac allograft vasculopathy and promoted regulatory T cell expansion. Scand J Immunol. 2013;78(1):44-49. 42. Zeng Q, Ng YH, Singh T, et al. B cells mediate chronic allograft rejection independently of antibody production. J Clin Invest. 2014;124(3):1052-1056. 43. Wehner JR, Fox-Talbot K, Halushka MK, Ellis C, Zachary AA, Baldwin WM,3rd. B cells and plasma cells in coronaries of chronically rejected cardiac transplants. Transplantation. 2010;89(9):1141-1148. 44. Gareau A, Hirsch GM, Lee TD, Nashan B. Contribution of B cells and antibody to cardiac allograft vasculopathy. Transplantation. 2009;88(4):470-477. 45. Millington TM and Madsen JC. Innate immunity and cardiac allograft rejection. Kidney Int Suppl. 2010;(119):S18-21. doi(119):S18-21. 46. Kitchens WH, Chase CM, Uehara S, et al. Macrophage depletion suppresses cardiac allograft vasculopathy in mice. Am J Transplant. 2007;7(12):2675-2682. 26 47. Huibers M, De Jonge N, Van Kuik J, et al. Intimal fibrosis in human cardiac allograft vasculopathy. Transpl Immunol. 2011;25(2-3):124-132. 48. Ankersmit HJ, Moser B, Roedler S, et al. Death-inducing receptors and apoptotic changes in lymphocytes of patients with heart transplant vasculopathy. Clin Exp Immunol. 2002;127(1):183-189. 49. Hirohashi T, Chase CM, Della Pelle P, et al. A novel pathway of chronic allograft rejection mediated by NK cells and alloantibody. Am J Transplant. 2012;12(2):313-321. 50. Uehara S, Chase CM, Colvin RB, Russell PS, Madsen JC. Further evidence that NK cells may contribute to the development of cardiac allograft vasculopathy. Transplant Proc. 2005;37(1):70-71. 51. Wehner J, Morrell CN, Reynolds T, Rodriguez ER, Baldwin WM,3rd. Antibody and complement in transplant vasculopathy. Circ Res. 2007;100(2):191-203. 52. Mitchell RN. Graft vascular disease: immune response meets the vessel wall. Annu Rev Pathol. 2009;4:19-47. 53. Devitt JJ, Rice A, McLean D, Murray SK, Hirsch GM, Lee TD. Impact of donor benign intimal thickening on cardiac allograft vasculopathy. J Heart Lung Transplant. 2013;32(4):454-460. 54. Atkinson C, Horsley J, Rhind-Tutt S, et al. Neointimal smooth muscle cells in human cardiac allograft coronary artery vasculopathy are of donor origin. J Heart Lung Transplant. 2004;23(4):427435. 55. Hollenberg SM, Klein LW, Parrillo JE, et al. Coronary endothelial dysfunction after heart transplantation predicts allograft vasculopathy and cardiac death. Circulation. 2001;104(25):30913096. 56. Minami E, Laflamme MA, Saffitz JE, Murry CE. Extracardiac progenitor cells repopulate most major cell types in the transplanted human heart. Circulation. 2005;112(19):2951-2958. 57. de Weger RA, Verbrugge I, Bruggink AH, et al. Stem cell-derived cardiomyocytes after bone marrow and heart transplantation. Bone Marrow Transplant. 2008;41(6):563-569. 58. Zheng Q, Liu S, Song Z. Mechanism of arterial remodeling in chronic allograft vasculopathy. Front Med. 2011;5(3):248-253. 59. Chen X, Kelemen SE, Autieri MV. AIF-1 expression modulates proliferation of human vascular smooth muscle cells by autocrine expression of G-CSF. Arterioscler Thromb Vasc Biol. 2004;24(7):1217-1222. 60. Autieri MV, Kelemen S, Thomas BA, Feller ED, Goldman BI, Eisen HJ. Allograft inflammatory factor-1 expression correlates with cardiac rejection and development of cardiac allograft vasculopathy. Circulation. 2002;106(17):2218-2223. 61. Song G, Fang Y, Wang X, Wu S, Song H. The effects of platelet-derived growth factor in rat cardiac allograft vasculopathy and fibrosis. Transplant Proc. 2008;40(8):2716-2719. 62. Tuuminen R, Nykanen AI, Krebs R, et al. PDGF-A, -C, and -D but not PDGF-B increase TGF-beta1 and chronic rejection in rat cardiac allografts. Arterioscler Thromb Vasc Biol. 2009;29(5):691-698. 27 63. Iida M, Tanabe K, Matsushima-Nishiwaki R, Kozawa O, Iida H. Adenosine monophosphateactivated protein kinase regulates platelet-derived growth factor-BB-induced vascular smooth muscle cell migration. Arch Biochem Biophys. 2013;530(2):83-92. 64. Raines EW. PDGF and cardiovascular disease. Cytokine Growth Factor Rev. 2004;15(4):237-254. 65. Khan R, Agrotis A, Bobik A. Understanding the role of transforming growth factor-beta1 in intimal thickening after vascular injury. Cardiovasc Res. 2007;74(2):223-234. 66. Booth AJ, Csencsits-Smith K, Wood SC, Lu G, Lipson KE, Bishop DK. Connective tissue growth factor promotes fibrosis downstream of TGFbeta and IL-6 in chronic cardiac allograft rejection. Am J Transplant. 2010;10(2):220-230. 67. Euler-Taimor G and Heger J. The complex pattern of SMAD signaling in the cardiovascular system. Cardiovasc Res. 2006;69(1):15-25. 68. Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13(8):952-961. 69. Chen PY, Qin L, Barnes C, et al. FGF regulates TGF-beta signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep. 2012;2(6):1684-1696. 70. Qin L, Huang Q, Zhang H, et al. SOCS1 prevents graft arteriosclerosis by preserving endothelial cell function. J Am Coll Cardiol. 2014;63(1):21-29. 71. Zhao DX, Hu Y, Miller GG, Luster AD, Mitchell RN, Libby P. Differential expression of the IFNgamma-inducible CXCR3-binding chemokines, IFN-inducible protein 10, monokine induced by IFN, and IFN-inducible T cell alpha chemoattractant in human cardiac allografts: association with cardiac allograft vasculopathy and acute rejection. J Immunol. 2002;169(3):1556-1560. 72. Cailhier JF, Laplante P, Hebert MJ. Endothelial apoptosis and chronic transplant vasculopathy: recent results, novel mechanisms. Am J Transplant. 2006;6(2):247-253. 73. Johnson A and DiPietro LA. Apoptosis and angiogenesis: an evolving mechanism for fibrosis. FASEB J. 2013;27(10):3893-3901. 74. Mendez-Fernandez YV and Major AS. Humanizing the problem of transplant vasculopathy. Arterioscler Thromb Vasc Biol. 2012;32(2):163-164. 75. Malone F, Carper K, Reyes J, Li W. gammadeltaT cells are involved in liver transplant tolerance. Transplant Proc. 2009;41(1):233-235. 28