Electronic Supplementary Material (a) (b) Fig. S1a (a) FT
advertisement
 (b) Fig. S1a (a) FT")
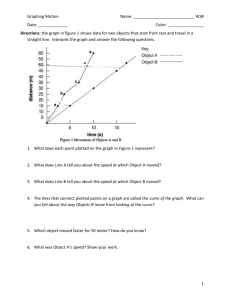
Electronic Supplementary Material (a) (b) Fig. S1a (a) FT-IR spectra of totally acetylated -Ala-Cellulose. The sample was prepared according to (Tezuka and Tsuchiya 1995). In brief for the sample preparation, -Ala-Cellulose 10 (DSO = 1.41, 0.5 g) was treated with a large excess amount of acetic anhydride/4-dimethylamino pyridine/absolute pyridine (distilled from KOH) mixture at 100 °C for 1.0 hr. The product was precipitated from anhydrous diethyl ether (distilled from sodium), and then, reprecipitated from chloroform–anhydrous diethyl ether. The purified sample (ca. 2.0 mg) was dissolved in chloroform and casted into a dried film on the single crystal NaCl prism (thickness, 3.0 mm). The FT-IR spectrum of the sample was recorded by means of the transmission mode (16 scans, curve 2) and compared with that of the Ala-Cellulose (curve 1). The acetylated (Ac)--Ala-Cellulose (Figure S1a, curve 2) exhibited absorption bands at 1,748 cm-1 (-ester carbonyls of O--Ala and O-Ac groups, the amide I at 1,649 cm-1, and the amide II at 1,561 cm-1 (N-Ac groups). While these absorption bands were not observed for -Ala-Cellulose • TFA 10 (curve 1). The disappearance of the broad O-H stretching vibration (3,500 cm-1, curve 2) with leaving the N-H stretching vibration for the Ac--Ala-Cellulose at 3,336 cm-1 (curve 1) indicated the total O- and N-acetylation of -Ala-Cellulose. (b) Carbonyl-signals region in the quantitative 13C-NMR spectrum of the totally acetylated -Ala-Cellulose 300 mg/mL CDCl3). The non NOE-gated decoupling plus program (factory preset name, C13IG) of a Bruker AVANCE400 instrument with several parameter modifications (duration, d1 = 40 sec; temperature, T = 40 °C) was employed, and the scan number was 1,500 times. As indicated earlier by (Tezuka and Tsuchiya 1995), the carbonyl signals of 6-OAc (170.0 ppm), 3-O-Ac (169.6 ppm), and 2-O-Ac (169.3 ppm) were observed in split. The N-Ac carbonyl on the Ala was assigned at 165.3 ppm). The signal-integration ratio of the four kinds of different carbonyls gives the regional DSO for -Ala was (6-OβAla, 3-OβAla, 2-OβAla) = (0.75, 0.40, 0.34) (total DSO = 1.49). Fig. S2 Gel permeation chromatography of the -Ala-Cellulose 10, deprotected conjugates N-[Boc(SerPO3H2)]-Ala-Cellulose 14, and N-[Boc(SerPO3H2)-Asp]--Ala-Cellulose 15, using a high performance liquid chromatography system (Hitachi High-technologies Lachrom Elite), which equipped with three columns connected in serial (a TSK guard column Super AW-H and a pair of TSKgelSuper AWM-H, calibrated with poly(ethylene oxide) standards; column temperature, 50 oC; flow rate, 0.5 mL/min; sample concentration, 0.10–0.15 wt.%). The mobile phase was a 10 mM LiBr/DMSO solution, and the elution profiles were monitored by a refractive index detector. The elution profiles indicated that the weight-average molecular weight, Mw, of the -Ala-Cellulose 10 was estimated as 56,100, while those of conjugates 14 and 15 were as 70,600 and 86,800, respectively, which indicate the parent cellulose back bones were retained mostly un-degraded during the deprotection treatment in the acidic media. Fig. S3 Phospho-amino acid analysis (PAA) chromatograms of the hydrolysates from N-[Ser(PO3H2)]--AlaCellulose 10 (solid line) and standard mixture (Ser(PO3H2), Asp, and -Ala, broken line). The hydrolysis and analytical protocols were given by (Capony and Demaille 1983) as follows: hydrolysis under reduced pressure at 110°C for 2 hr for both of the sample and standard. Note that the protocol can detect only the O-phospho-amino acids, and in those cases of the sample and standard, the Ser(PO 3H2) was found at the retention time of 6.47 min. The standard amount fed was same to the expected content of Ser(PO 3H2) in N-[Ser(PO3H2)]- -Ala-Cellulose 11 (calc), based on the DSO and DSN values. The results were 2.28 µmol/mg for calc and 2.27 ± 0.02 µmol/mg for found, indicating that no elimination of the phosphoryl groups from the Ser(PO 3H2) during the one-pot deprotection.