biochem ch 39 [10-2
advertisement

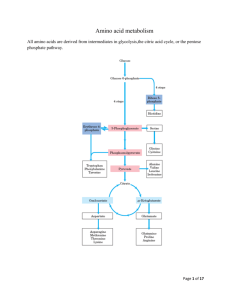
Biochem Chapter 39 Synthesis and Degradation of Amino Acids All states enacted legislation requires newborns be screened for various metabolic abnormalities, of which PKU is one o Guthrie assay – Bacillus subtilis plated on agar plate containing β2-thienylalanine (inhibitor of B. subtilis growth); blood sample obtained from infant (dried blood spot on filter disk) and placed on agar plate If phenylalanine content of blood is greater than 2-4 mg/dL, phenylalanine will counteract effects of β2-thienylalanine, and bacterial growth will occur o Alternative assay is microfluorometric determination of phenylalanine, via incorporation into ninhydrincopper complex with dipeptide L-leucyl-L-alanine o Positive results with either assay require verification and actual measurement of phenylalanine levels, using either HPLC separation of components of blood or enzymatic and fluorometric assays Role of Cofactors in Amino Acid Metabolism Amino acid metabolism requires 3 important cofactors o Pyridoxal phosphate (PLP) is quintessential coenzyme of amino acid metabolism (made from vitamin B6) o Coenzyme tetrahydrofolate (FH4) is required in certain amino acid pathways to either accept or donate a one-carbon group o Coenzyme tetrahydrobiopterin (BH4) required for ring hydroxylations Reactions involve molecular oxygen, and one atom of oxygen incorporated into product Second found in water BH4 important for synthesis of tyrosine and neurotransmitters Amino Acids Derived from Intermediates of Glycolysis Serine – in biosynthesis of serine from glucose, 3-p[hosphoglycerate first oxidized to 2-keto compound, which is then transaminated to form phosphoserine o Phosphoserine phosphatase removes phosphate, forming serine o Major sites of serine synthesis liver and kidney o Can be used by many tissues and generally degraded by transamination to hydroxypyruvate, followed by reduction and phosphorylation to form 2-phosphoglycerate (intermediate of glycolysis that forms phosphoenolpyruvate (PEP) and, subsequently, pyruvate o Can undergo β-elimination of hydroxyl group, catalyzed by serine dehydratase, to form pyruvate directly o When serine levels fall, serine synthesis increased by induction of 3-phosphoglycerate dehydrogenase and release of feedback inhibition of phosphoserine phosphatase (caused by higher levels of serine) When serine levels rise, synthesis of serine decreases because synthesis of dehydrogenase repressed and phosphatase inhibited Glycine – synthesized from serine (major pathway) or threonine (minor pathway) o Conversion from serine by reversible reaction that involves FH4 (derived from folate) and PLP o Conversion from threonine requires threonine degradation (aldolase-like reaction because threonine contains hydroxyl group located 2 carbons from carbonyl group) o Oxalate (produced from glycine or obtained from diet) forms precipitates with calcium; kidney stones (renal calculi) often composed of calcium oxalate Lack of transaminase that can convert glyoxylate to glycine leads to primary oxaluria type I (PH 1), which has a consequence of renal failure attributable to excessive accumulation of oxalate in kidney o Conversion of glycine to glyoxylate by enzyme D-amino acid oxidase – degradative pathway of glycine Once glyoxylate formed, it can be oxidized to oxalate (sparingly soluble and tends to precipitate in kidney tubules, leading to formation of kidney stones) 40% of oxalate formation in liver comes from glycine metabolism Dietary oxalate accumulation is low contributor to excreted oxalate in urine because of poor absorption of oxalate in intestine o Glyoxalate can be transaminated back to glycine o Generation of energy from glycine occurs through a dehydrogenase (glycine cleavage enzyme) that oxidizes glycine to CO2, NH3, and carbon that is donated to FH4 Cysteine – carbons and nitrogen for cysteine synthesis provided by serine, and sulfur provided by methionine o Serine reacts with homocysteine (produced from methionine) to form cystathionine (catalyzed by cystathionine β-synthase) cleavage of cystathionine by cystathionase produces cysteine and αketobutyrate, which forms succinyl-CoA via propionyl-CoA Both cystathionine β-synthase (β-elimination) and cystathionase (γ-elimination) require PLP o Cystathioninuria – presence of cystathionine in urine; relatively common in premature infants As infants mature, cystathionase levels rise, and levels of cystathionine in urine decrease In adults, genetic deficiency of cystathionase causes cystathioninuria Individuals with genetically normal cystathionase can develop cystathioninuria from dietary deficiency of pyridoxine (vitamin B6) because cystathionase requires cofactor PLP No characteristic clinical abnormalities observed in cystathionase deficiency o Cystinuria – caused by defect in transport protein that carries cystine, lysine, arginine, and ornithine into intestinal epithelial cells and permits resorption of these AAs by renal tubular cells Cystine not very soluble in urine; forms renal caliculi o Cystinosis – rare disorder caused by defective carrier that normally transports cystine across lysosomal membrane from lysosomal vesicles to cytosol Cystine accumulates in lysosomes in many tissues and forms crystals, leading to depletion of intracellular cysteine levels Children with this disorder develop renal failure by 6-12 years of age o When cysteine degraded, nitrogen converted to urea, carbons to pyruvate, and sulfur to sulfate Sulfate generation in aqueous medium essentially generating sulfuric acid, and both acid and sulfate need to be disposed of in urine Sulfate used in most cells to generate activated form of sulfate PAPS, which is used as sulfate donor in modifying carbs or AAs in various structures (GAGs) and proteins in body o Conversion of methionine to homocysteine and homocysteine to cysteine is major degradative route for these 2 AAs; this is the only degradative route for homocysteine, so vitamin B6 deficiency or congenital cystathionine β-synthase deficiency can result in homocystinemia, which has been reported to be associated with cardiovascular disease Alanine – produced from pyruvate by transamination reaction catalyzed by alanine aminiotransaminase (ALT) and may be converted back to pyruvate by reaction reversal o Major gluconeogenic AA because it’s produced in many tissues for transport of nitrogen to liver Amino Acids Related to TCA Cycle Intermediates Amino acids through α-ketoglutarate o Glutamate – derived from α-ketoglutarate either y transamination or by glutamate dehydrogenase reaction; because α-ketoglutarate can be synthesized from glucose, all carbons of glutamate can be obtained from glucose When glutamate degraded, it is converted back to α-ketoglutarate either by transamination or glutamate dehydrogenase In liver, α-ketoglutarate leads to formation of malate, which produces glucose via gluconeogenesis, so glutamate can be derived from glucose or can be turned into glucose Glutamate used for synthesis of glutamine, proline, ornithine, and arginine, as well as providing glutamyl moiety of glutathione, which is an important antioxidant o Glutamine – produced from glutamate by glutamine synthetase, which adds NH4+ to carboxyl group of side chain, forming an amide (one of only 3 enzymes that can fix free ammonia into organic molecule; other 2 are glutamate dehydrogenase and carbamoyl phosphate synthetase I) Glutamine reconverted to glutamate by glutaminase, which is particularly important in kidney Ammonia it produces enters urine and can be used as expendable cation to aid in excretion of metabolic acids o Proline – glutamate is phosphorylated then converted to glutamate 5-semialdehyde by reduction of side-chain carboxyl group to aldehyde Semialdehyde spontaneously cyclizes (forming internal Schiff base between aldehyde and αamino group); reduction of cyclic compound yields proline Hydroxyproline formed only after proline incorporated into collagen by prolyl hydroxylase system, which uses molecular oxygen, iron, α-ketoglutarate, and ascorbic acid Proline converted back to glutamate semialdehyde, which is oxidized to form glutamate Synthesis and degradation of proline use different enzymes even though same intermediates Hydroxyproline has entirely different degradative pathway; presence of hydroxyl group in hydroxyproline allows aldolase-like reaction to occur once ring has been hydrolyzed, which isn’t possible with proline o Arginine – synthesized from glutamate via glutamate semialdehyde, which is transaminated to form ornithine (intermediate of urea cycle); activity of ornithine aminotransferase greatest in epithelial cells of small intestine Reactions of urea cycle produce arginine from ornithine; quantities of arginine generated by urea cycle adequate only for adult and are insufficient to support growth, so during periods of growth, arginine is essential amino acid If arginine used for protein synthesis, levels of ornithine will drop, thereby slowing urea cycle, stimulating formation of ornithine from glutamate Arginine cleaved by arginase to form urea and ornithine; if ornithine present in amounts in excess of those required for urea cycle, it is transaminated to glutamate semialdehyde, which is reduced to glutamate; conversion of aldehyde to primary amine requires PLP o Histidine – cannot be synthesized in human body, but it can form glutamate when it is degraded Histidine converted to FIGLU, and subsequent reactions transfer one carbon of FIGLU to FH4 pool and release NH4+ and glutamate Amino acids related to oxaloacetate o Aspartate produced by transamination of oxaloacetate; reaction readily reversible o Asparagine – formed from aspartate by reaction in which glutamine provides nitrogen for formation of amide group; reaction catalyzed by asparaginase, which hydrolyzes asparagine to NH4+ and aspartate (analogous to reaction catalyzed by glutaminase) Certain types of tumor cells (particularly leukemic cells) require asparagine for growth Asparaginase can be used as antitumor agent, converting asparagine to aspartate in blood, decreasing amount of asparagine available for tumor cell growth Amino acids that form from fumarate o Aspartate – major route for aspartate degradation involves conversion to oxaloacetate, but carbons from aspartate can form fumarate in urea cycle, generating cytosolic fumarate, which must be converted to malate (using cytoplasmic fumarase) for transport into mitochondria for oxidative or anaplerotic purposes Analogous sequence of reactions occurs in purine nucleotide cycle; aspartate reacts with IMP to form adenylsuccinate that is then cleaved, forming AMP and fumarate o Phenylalanine – converted to tyrosine by hydroxylation reaction o Tyrosine – produced from phenylalanine or obtained from diet, is oxidized, ultimately forming acetoacetate and fumarate Oxidative steps required to reach this point not energy generating Conversion of fumarate to malate, followed by action of malic enzyme, allows carbons to be used for gluconeogenesis Amino acids that form succinyl-CoA o Essential amino acids methionine, valine, isoleucine, and threonine degraded to form propionyl-CoA; conversion of propionyl-CoA to succinyl-CoA common to their degradative pathways Propionyl-CoA also generated from oxidation of odd-chain fatty acids Propionyl-CoA carboxylated in reaction that requires biotin and forms D-methylmalonyl-CoA, which is racemized to L-metylmalonyl-CoA, which is rearranged in vitamin B12-requiring reaction to produce succinyl-CoA (TCA cycle intermediate) o Methionine – converted to S-adenosylmethionine (SAM), which donates its methyl group to other compounds to form S-adenosylhomocysteine (SAH) SAH converted to homocysteine Methionine regenerated from homocysteine by reaction that requires FH4 and vitamin B12 By reactions that require PLP, homocysteine can provide sulfur needed for synthesis of cysteine Carbons of homocysteine metabolized to α-ketobutyrate, which undergoes oxidative decarboxylation to propionyl-CoA, which is then converted to succinyl-CoA o Threonine – primarily degraded by PLP-requiring dehydratase to NH3 and α-ketobutyrate, which subsequently undergoes oxidative decarboxylation to form propionyl-CoA o Branched-chain amino acids (valine, isoleucine, and leucine) are universal fuels, and degradation of amino acids occurs at low levels in mitochondria of most tissues, but muscle carries out highest level of BCAA oxidation BCAAs make up 25% of content of average protein, so their use as fuel is significant Degradative pathway for valine and isoleucine generates energy and provides precursors to replenish TCA cycle intermediates (anaplerosis) Valine and isoleucine contain carbons that form succinyl-CoA Initial step of degradation of BCAAs is transamination reaction; enzyme that catalyzes this reaction present in most tissues, but activity particularly high in muscle Second step of degradation pathway, α-keto analogs undergo oxidative decarboxylation by αketo acid dehydrogenase complex; highest level of activity for this is in muscle tissue Pathways for degradation of BCAAs follow parallel routes and are analogous to β-oxidation of fatty acids, so NADH and FAD(2H) generated for energy production Thiamine deficiency leads to accumulation of α-keto acids in blood because of inability of pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and branched-chain α-keto acid dehydrogenase to catalyze reactions; can be caused by chronic alcoholism Valine and isoleucine converted to succinyl-CoA Isoleucine also forms acetyl-CoA Leucine forms acetoacetate and acetyl-CoA and is strictly ketogenic o In maple syrup urine disease, branched-chain α-keto acid dehydrogenase that oxidatively decarboxylates BCAAs is defective BCAAs and their α-keto analogs (produced by transamination) accumulate and appear in urine, giving it odor of maple syrup or burnt sugar Accumulation of α-keto analogs leads to neurologic complications Condition difficult to treat by dietary restriction because abnormalities in metabolism of 3 essential AAs contribute to disease Amino Acids that Form Acetyl-CoA and Acetoacetate Phenylalanine – converted to tyrosine, which undergoes oxidative degradation; last step in pathway produces both fumarate and ketone body acetoacetate o Deficiencies of different enzymes in pathway result in PKU, tyrosinemia, and alkaptonuria o Alkaptonuria – occurs when homogentisate (intermediate in tyrosine metabolism) can’t be further oxidized because next enzyme in pathway (homogentisate oxidase) defective Homogentisate accumulates and auto-oxidizes, forming dark pigment, which discolors urine and stains diapers of affected infants Later in life, chronic accumulation of pigment in cartilage may cause joint pain o Phenylalanine – hydroxylated to form tyrosine by mixed-function oxidase (PAH) which requires molecular oxygen and BH4 BH4 converted to quinonoid dihydrobiopterin by above reaction BH4 can be synthesized in body from GTP, but body contains limited amounts, so dihydrobiopterin must be reconverted to BH4 for reaction to continue to produce tyrosine o Transient tyrosinemia – frequently observed in newborns, especially those that are premature Condition benign, and dietary restriction of protein returns plasma tyrosine levels to normal Biochemical defect is low level of 4-hydroxyphenylpyruvate dioxgenase due to immaturity Enzyme requires ascorbate, so ascorbate supplementation aids in reducing circulating tyrosine levels o Tyrosinemia II caused by genetic deficiency of TAT and may lead to lesions of eye and skin as well as neurologic problems; patients treated with low-tyrosine, low-phenylalanine diet o Tyrosinemia I (AKA tyrosinosis) caused by genetic deficiency of fumarylacetoacetate hydrolase Acute form associated with liver failure, cabbage-like body odor, and death in first year of life o Small subset of patients with hyperphenylalaninemia show appropriate reduction in plasma phenylalanine levels with dietary restriction of phenylalanine, but still develop progressive neurologic symptoms and seizures and usually die within first 2 years of life (“malignant” hyperphenylalaninemia) Infants exhibit normal PAH activity but have deficiency in DHPR (enzyme required for regeneration of BH4, which is a cofactor of PAH) Less frequently, DHPR activity normal but defect in biosynthesis of BH4 exists Dietary therapy corrects hyperphenylalaninemia, but BH4 is cofactor for 2 other hydroxylations required in synthesis of neurotransmitters in brain (hydroxylation of tryptophan to 5hydroxytryptophan and hydroxylation tyrosine to L-dopa) Resulting deficit in CNS neurotransmitter activity at least in part responsible for neurologic manifestations and eventual death of patients Tryptophan – oxidized to produce alanine (from nonring carbons), formate, and acetyl-CoA o Tryptophan both glucogenic and ketogenic o NAD+ and NADP+ produced from ring structure of tryptophan, so tryptophan spares dietary requirement for niacin o The higher the dietary levels of tryptophan, the lower levels of niacin required to prevent symptoms of deficiency o If dietary levels of niacin and tryptophan insufficient, pellagra results Symptoms of pellagra are dermatitis, diarrhea, dementia, and death o Abnormal metabolism of tryptophan occurs in vitamin B6 deficiency Kynurenine intermediates in tryptophan degradation can’t be cleaved because kynureninase requires PLP derived from vitamin B6, so intermediates enter minor pathway for tryptophan metabolism and produce xanthurenic acid, which is excreted in urine Threonine – major route of degradation is by threonine dehydratase; in minor pathway, threonine degradation by threonine aldolase produces glycine and acetyl-CoA in liver Isoleucine – produces both succinyl-CoiA and acetyl-CoA Leucine – purely ketogenic and produces HMG-CoA, which is cleaved to form acetyl-CoA and ketone body acetoacetate; most tissues in which it is oxidized can use ketone bodies with exception of liver o Leucine is universal fuel, with primary metabolism occurring in muscle Lysine – can’t be directly transaminated at either of its 2 amino groups; degraded by complex pathway in which saccharopine, α-ketoadipate, and crotonyl-CoA are intermediates o During degradation pathway, NADH, and FADH2 generated for energy o Ultimately, lysine generates acetyl-CoA and is strictly ketogenic Phenylketonuria Classic PKU – caused by mutations in gene located on chromosome 12 that encodes enzyme PAH o PAH normally catalyzes hydroxylation of phenylalanine to tyrosine (rate-limiting step in major pathway by which phenylalanine catabolized); single base substitution in gene is cause of this PKU is heterogeneous phenotype with varying degrees of hyperphenylalaninemia and different types of mutations