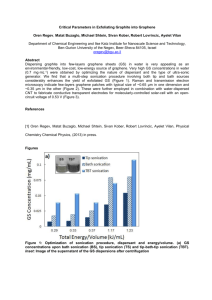
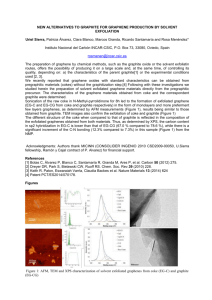
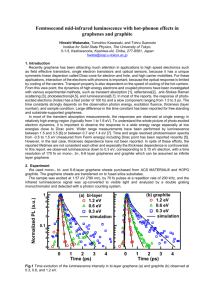
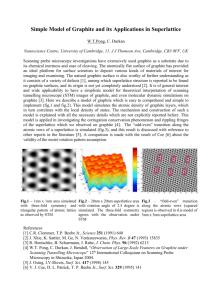
Graphene Reinforced Carbon Composite Foams with Improved
advertisement

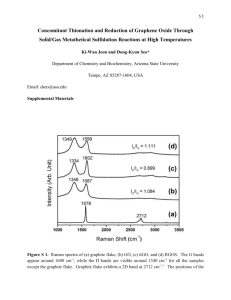
Graphene Reinforced Carbon Composite Foams with Improved Strength and EMI Shielding from Sucrose and Graphene Oxide R. Narasimman*, Sujith Vijayan, and K. Prabhakaran Department of Chemistry, Indian Institute of Space Science and Technology, Thiruvananthapuram- 695 547, India 1. Preparation of Graphene Oxide The concentrated sulfuric acid (98%), concentrated hydrochloric acid (36%), potassium permanganate (99.5%), sodium hydroxide (96%), and hydrogen peroxide (30%) used were analytical reagent grade and purchased from Merck India, Mumbai. All reagents were used without further purification. Graphene oxide (GO) was prepared from graphite powder using the procedure reported by Aboutalebiet al.1, 2 Graphite powder (5 g) and concentrated sulfuric acid (98%, 1 liter) were mechanically stirred using Teflon paddle for 2 h in a three-neck round bottom flask fitted with a water condenser. The temperature of the bath was maintained at 25 oC during the stirring. 50 g KMnO4 was added lot wise to the mixture in 1 h so as to maintain the temperature of the reaction mixture below 50 oC. The temperature of the reaction mixture was then increased to 50 oC and maintained for 24 h with continuous vigorous stirring. One liter of distilled water was added after cooling the reaction mixture to a temperature of < 5 oC in an ice bath and then stirred for 1 h. 25 ml of 30 vol.% H2O2 was added to the reaction mixture to obtain a brown colloidal solution. The brown colloidal solution was further stirred for 1 h and then centrifugally washed three times with 1M HCl solution. Further, the colloidal solution was centrifugally washed several times with distilled water until the pH reaches between 6 and 7. Finally, the water present in the GO solution was exchanged with acetone by repeated centrifugation and decanting. The GO concentration in the acetone solution was gravimetrically estimated as 4 mg.ml-1. 2. Characterization of GO A Fourier transform infrared (FTIR) spectrum of the GO was recorded using a FTIR spectrophotometer (Spectrum-150, PerkinElmer, USA) in the spectral range of 4000 to 400 cm-1. Thermogravimetric analysis (TGA) of the GO and solid organic foam samples was carried out in nitrogen atmosphere using a thermogravimetric analyzer (Q-50, TA instruments, USA) at a heating rate of 2 oC min-1. X-Ray diffraction (XRD) measurements of the GO and graphite were carried out in an X-ray diffractometer (X’pert Pro, Philips, USA) using Cu K radiations (l = 1.54056 Å). The diffraction patterns were recorded at 2 values in the range of 10 to 80o with a step size of 0.07o using monochromatic X-rays. Raman spectra were taken on a RAMAMLOG 6 (Spex, USA) using a 50 x objective lens with a 514.5 nm laser excitation. The atomic force microscope (AFM, Agilent, USA) was used to observe the topography of the GO. 50 mg of GO solution was drop casted on a freshly cleaved mica sheet and dried for two days in ambient condition. The images were taken using tapping mode (AAC mode). A Si3N4 tip was used for the imaging. 3. Characterization of GO dispersion in sucrose 0.5 g of GO-sucrose powders containing various concentrations of GO were dissolved in 300 l distilled water. The dispersions drop-casted on a glass slide is immediately covered with a cover glass and observed under an optical microscope in transmission mode (Leica microsystem, Wetzlar, Germany). Figures: Figure S1. Thermogram of the GO Figure S2. FTIR spectrum of GO Figure S3. XRD spectra of the graphite and GO Figure S4. Raman spectra of the graphite and GO Figure S5. AFM image and height profile of GO sample Figure S1 shows the TGA of GO. The main weight loss of GO happens in the temperature range of 150 to 200 oC due to the decomposition of the liable oxygen functional groups introduced during oxidation of graphite.3 Figure S2 shows the FTIR spectrum of GO showing the presence of various functional groups. The peaks at 3394, 1717, 1232 and 1028 cm-1are corresponding to –OH stretching vibrations, -C=O stretching vibrations, -C-OH stretching vibrations and -C-O-C- stretching vibrations, respectively.3 The XRD spectra of graphite and GO are shown in Figure S3. The XRD pattern of graphite shows a sharp peak at 26.2o, which is due to the reflection from (002) plane. Whereasthe sharp peak in the XRD pattern of graphite vanishes after the oxidation treatmentand a broad peak observed at a 2 value of 12o, corresponding to a d-spacing of 7.4 Å. This d-spacing represents, approximately, one-molecularlayer thick water trapped between the GO layers.1 Figure S4 shows the Raman spectra of the graphite and GO. The graphite shows sharp peaks at 1582 and 2745 cm-1 corresponding to G band and 2D band, respectively. On the other hand, Raman spectrum of GO shows peaks at 1368 and 1617 cm-1 corresponding to the D and G bands, respectively. The G-band is due to Raman active sp2 hybridized carbon atoms, and D-band is due to the defect that causes double resonance Raman scattering. The shift in G band from 1581 to 1617 cm-1is because the isolated double bonds in GO resonate at higher frequencies than that of graphite.1, 2 The GO sheets used are having irregular morphology with the average lateral sheet size of 12 m as calculated from the AFM images. The AFM image of the GO sheets and the height-profile are shown in Figure S5. The AFM image clearly reveals the single layer GO. The height-profile shows the thickness of GO sheet as approximately 0.9 nm.The thickness obtained is corresponding to the single layer graphene oxide as reported in the literature.1, 2 Figure S6. GO dispersions in sucrose solution at the GO concentrations of (a) 0.25 wt.% (b) 0.75 wt.% and (c) 1.25 wt.% (Agglomeration at GO concentration beyond 0.75 wt.% is clear from the optical image) Figure S7. Schematic of the EMI shielding measurements set up Figure S8. Total SE of the graphene reinforced carbon composite foams prepared at various GO concentrations References 1. S. H. Aboutalebi, A. T. Chidembo, M. Salari, K. Konstantinov, D. Wexler, H. K. Liu and S. X. Dou, Energy & Environmental Science, 2011, 4, 1855-1865. 2. S. H. Aboutalebi, M. M. Gudarzi, Q. B. Zheng and J.-K. Kim, Advanced Functional Materials, 2011, 21, 2978-2988. 3. J. I. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2008, 24, 10560-10564.