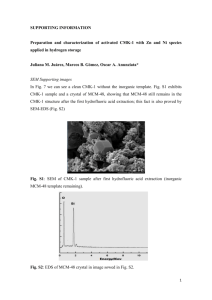
Supplemental_Material
advertisement

Supplemental Material
A. Corroboration of IGCM results by the MDGC method and Chain Free Energy
The incremental chemical potential was calculated by both Ideal Gas (IG) and Mean
Density (MD) methods.32 In the MD method, the incremental chemical potential for a chain of
length N is calculated from a simple average of sampled incremental chemical potentials over the
course of a simulation. The MD method assumes the ‘mean density’ of monomers in the gauge
cell reflects the simulation’s equilibrium state.
The function 𝜇𝑖𝑛𝑐𝑟 (𝑁) is then ‘built up’ by
averaging over many iterations of the same simulation for each value of N desired. In the IG
method, a histogram (series of 𝜇𝑖𝑛𝑐𝑟 values for Ni near N) of the incremental chemical potential
is collected. Histograms for different chain lengths are then combined to create a ‘smooth’
function 𝜇𝑖𝑛𝑐𝑟 (𝑁). The latter method requires a relatively fine grid of N, to ensure that the
histogram peaks overlap sufficiently to render a statistically sound average. The penalty of
utilizing a fine grid however is more than compensated for when compared to the MD method,
which takes anywhere from 20 to 100 simulations (per N) to render statistically sound results. In
order to test the applicability of both of these methods, two sets of IGCM simulations were
performed, one with MD and one with IG.
The MD simulations consisted of 20 to 100
simulations at each value of N = 25, 50, 100, 150 and 200 for each value of U = 0, -3, -5, -6…10. The IG simulations consisted of one simulation at each value of N = 3, 5, 10, 15…200, for
the same range of U values. Each set consisted of 400 equilibration and 500 production sets of
between 850,000 and 1 million Monte Carlo moves per set.
FIG. A.1. Dependence of the incremental chemical potential on the adsorption energy U for tethered chains of
different length N from 25 to 200. The intersection point corresponds to the CPA at U=Uc=-5.4 +/- 0.05. Free
jointed LJ chain model. Calculations with the incremental gauge cell MC method; (a) – MD, (b) – IG.. For the MD,
N = 25, 50, 100, 150, 200. For the IG, results are presented for N = 30, 40, …, 200. In both cases, there is clear
intersection at the point Uc = -5.4 +/- 0.05.
The chain incremental chemical potential for sufficiently large chains (N > 30) is shown
to be a constant in our work. It follows that chain free energy, which is the sum of the chain
incremental chemical potentials, should be linear in N for the same range of N. The linearity of
the chain free energy dependence on N is illustrated in Fig. A.2 below.
FIG. A.2. The chain free energy as a function of N. These calculations support the chain increment ansatz: for
sufficiently large N the incremental chemical potential is constant and the chain free energy in a linear function of N.
B. Geometrical method for determination of the CPA
As noted in the paper, DeGennes defined the CPA from the criterion that below the CPA,
the probability Pa of adsorption for a monomer distanced from the tethered end by n monomers
decreases to 0 as n increases. Above the CPA, this probability approaches a finite limit as 𝑛 →
∞. The respective graph is shown in Fig. B.1. (a). Likewise, utilizing the ansatz (2), the CPA
may be found from the extrapolation of the linear region of a plot relating the fraction of
adsorbed monomers to the adsorption energy Fig. B.1 (b).
FIG. B.1. (a) Probability of adsorption for a monomer distanced from the tethered end by n monomers, Pa;
Adsorption energies U = -3, -5.4 (CPA), -7, -8, and -10. Results averaged for 40 chains. (b) Fraction of adsorbed
monomers M/N as a function of adsorption energy -U; N = 200.
Futhermore, in the geometrical framework several important scaling relationships for
large chains have been developed. We are concerned here only with those governing the radii of
gyration Rx2, Ry2 and Rz2, from which are derived the radii of gyration normal and parallel to the
surface of the adsorbent:
2
𝑅𝑔⊥
= 𝑅𝑧2
2
𝑅𝑔∥
=
2
𝑅𝑥2 +𝑅𝑦
2
(B.1)
(B.2)
These scaling relationships make use of the crossover exponent 𝜙, and assume the following
forms:
2
𝑅𝑔⊥
𝑁𝜈
𝑐⊥1 𝑥 → ∞
𝑥=0
= ℎ⊥ (𝑥) = { 𝑐⊥2
𝜈3
−
|𝑥| 𝜙 𝑥 → −∞
2
𝑅𝑔∥
𝑁𝜈
𝑐∥1 𝑥 → ∞
𝑐∥2 𝑥 = 0
= ℎ∥ (𝑥) = { 𝜈 −𝜈
2 3
|𝑥| 𝜙
(B.3a-b)
𝑥 → −∞
In (B3.a-b) ν3 = 0.588 and ν2 = ¾ are the 3d and 2d Flory exponents for polymer chains20 and
𝑥 = 𝜏𝑁 𝜙 .
It is worth noting that these scaling equations are plausible qualitative
approximations, which reflect the asymptotic behavior at large N. Equations (B3.a-b) imply that
the ratio of these primary radii of gyration gives rise to a function that is invariant in N at the
critical point. Therefore, a plot of the ratio of the radii of gyration as a function of U for several
values of N should intersect at the critical point, Uc. This property is used to estimate the critical
point in the geometrical method. However, since it holds only with the provision of large enough
N, the geometrical method applied for finite length chains may overestimate the critical point, as
shown below in Fig. B.2.
FIG. B.2. Ratio of the radii of gyration of a tethered chain as a function of adsorption energy. Clear intersection
occurs at around U = -6.5, somewhat more negative in value than the IG/MD results.
For a long chain free in solution, the radii of gyration in each coordinate should approach
the same value, and thus the polymer should approach a sphere-like shape. The ratio of these
radii should therefore be unity, as is shown within experimental error in Fig. B.2. For a tethered
chain, this ratio will necessarily be slightly skewed, due to anisotropy of the chain and the
limitation of conformations thus imposed upon it. However, as the chain becomes large, this
anisotropy should decrease. Indeed, we see in Fig. B.2 that for chains beyond 50 monomers, this
effect is about the same. In the presence of an adsorption field, the tethered chains undergo a
geometrical transition, from the three-dimensional solvated chain, to one which is mostly
adsorbed and two-dimensional.
As implied by (B3.a-b), at the CPA, these curves should
intersect for all values of N. Fig. B.2. shows that this transition does indeed occur, though at a
value of U slightly more negative than found by the adsorption method. The geometrical CPA
occurs at approximately UcGM = -6.5 +/- 0.1.
In the repulsion limit at 𝑥 = 𝜏𝑁 𝜙 → ∞, both 𝑅⊥ and 𝑅∥ scale as 𝑁 𝜈3 , and respectively
𝑅⊥ /𝑅∥ ~1. At the very CPA in the limit of |𝜏| ≪ 𝑁 −1/2, ℎ⊥ and ℎ∥ approach certain constants,
ℎ⊥ (0) and ℎ∥ (0). Thus, the scaling ansatz (3) implies that 𝑅⊥ /𝑅∥ is independent of N at 𝜏 → 0.
This conclusion was used14-15,
20
for the practical calculation of the CPA from the point of
intersection of 𝑅⊥ /𝑅∥ versus U plots for the chains of different length N. However, since the
scaling ansatz holds only with the provision of large N, such a geometrical method applied for
finite length chains may overestimate the critical point. In addition it is worth mentioning, that
from the asymptotes (4) it follows that (𝑅⊥ /𝑅∥ )𝑁 𝜈2 ∝ |𝜏|
−𝜈2
𝜙
= |𝜏|−3/2 at 𝑥 → −∞. As such, the
CPA can be estimated by plotting (𝑅⊥ /𝑅∥ )𝑁 𝜈2 versus U and fitting to the asymptote |𝜏|−3/2.
However, it does not seem to be practical for the relatively short chains N<200, as seen from Fig.
B.3.
3
FIG. B.3. Plot of ratio of radii of gyration times 𝑁 𝜈2 versus adsorption energy. Asymptotic lines for |𝜏|−2 versus U
also included for Uc = -5.4 and -6.5.
C. Alternative consideration of the scaling at CPA.
𝑅⊥ is determined by the characteristic length l of the loops formed by non-adsorbed
𝑁
monomers, as 𝑅⊥ ∝ 𝑙 𝜈3 . The loop length 𝑙 ∝ 𝑀 ∝ 𝑁1−𝜙 , and, as such, 𝑅⊥ ∝ 𝑁 (1−𝜙)𝜈3 . 𝑅∥ is
determined by the distribution of M=𝑁 𝜙 adsorbed monomers; this distribution can be viewed as
resulting from a 2d random walk with the characteristic step equal to the characteristic loop
extension in the parallel direction, which should be of the order of 𝑅⊥ . Thus, (𝑅⊥ /𝑅∥ ) ∝ 𝑁 −𝜙𝜈̃2 ,
where 𝜈̃2 is the respective 2d random walk exponent. It is debatable whether this 2d random
walk should be treated as ideal or as a self-avoiding trajectory; in the former case 𝜈̃2 =1/2 and
(𝑅⊥ /𝑅∥ ) ∝ 𝑁 −1/4, while in the latter case 𝜈̃2 =3/4 and (𝑅⊥ /𝑅∥ ) ∝ 𝑁 −3/8. This consideration
negates the above conclusion that that 𝑅⊥ /𝑅∥ is independent of N at 𝜏 → 0, which is worth
additional verification.