lec06_2013 - Andrew.cmu.edu
advertisement

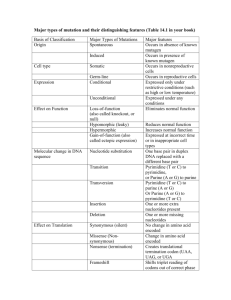
Biochemistry Lecture 6 January 24, 2013 Lecture 6: UV Absorption of Amino Acids & Protein Structure Assigned reading: Horton - 3.2B, 3.9-3.10. Key Terms: UV Absorption Peptide/protein nomenclature Edman degradation Cyanogen bromide cleavage Nelson 5e - box 3.1, 3.4. OLI: Quiz: 2 Chymotrypsin & trypsin cleavage Protein sequence determination. + 6A. UV Absorption Properties of Amino Acids H3N Three aromatic amino acids absorb light in the ultraviolet range (UV). O O N H + H3N O O OH + H3N O The extinction coefficients (or molar absorption coefficients) of these amino acids are: 5 Extinction Coefficient (MAX) 5,500 M-1cm-1 (280 nm) 1,490 M-1cm-1 (274 nm~280nm) 220 M-1cm-1 (257 nm) The amount of light absorbed by a solution of concentration [X] is given by the Beer-Lambert Law: 4 3 2 1 0 I A log O [ X ] l I where A is the absorbance of the sample; I0 is the intensity of the incident light; I is the intensity of the light that leaves the sample. 6 Absorbance Amino acid Trp Tyr Phe O 230 240 250 260 270 280 290 300 310 320 330 Wavelength (nm) ε is the molar extinction coefficient at a specific wavelength, e.g. at max; [X] is the concentration of the absorbing species l is the path length (usually 1 cm). Therefore, given a known extinction coefficient it is possible to measure the concentration of a protein. Calculation of molar extinction coefficients: If a molecule contains a mixture of N different chromophores, the molar extinction coefficient can generally be calculated as the sum of the molar extinction coefficient for each absorbing group in the protein: N Protein i i 1 Therefore, the molar extinction coefficient for a protein can be calculated from its amino acid composition. Example: A protein has two Tryptophan (Trp) residues and one Tyrosine (Tyr) what is its extinction coefficient? 1 Biochemistry Lecture 6 January 24, 2013 6B. Protein Structural Hierarchy: 1. Primary structure (1°): The amino acid sequence. 2. Secondary structure (2°): Configuration of mainchain. 3. Tertiary structure (3°): Side chain packing in the 3-D structure. 4. Quaternary structure (4°): Association of subunits. O 6C. Primary Structure H3C CH3 O H CH3 O H N H3N O N O + H H H O O H O O CH3 H 3N + O O H N O N H O O H 3C CH3 Example: Draw the structure of Gly-Ala-Ala i) draw mainchain atoms first. ii) add sidechains Sequencing Proteins Protein Sequencing: Edman Degradation We will focus on N-terminal sequencing using Edman degradation coupled with fragmentation of the peptide in the case of larger proteins. Edman Degradation: Cleaves single amino acids from the amino-terminus. Producing: i) The PTH derivative of the released amino acid can be identified. ii) A peptide that is one residue shorter is produced, this can be treated with PITC to obtain the next residue, etc. iii) Errors accumulate because the release of the PTH-amino acid is not 100%, limiting the length of sequence to ~75 residues: AGCTWGCAYPF AGCTWGCAYPF 100% Ala (A) GCTWGCAYPF CTWGCAYPF GCTWGCAYPF CTWGCAYPF AGCTWGCAYPF GCTWGCAYPF CTWGCAYPF AGCTWGCAYPF GCTWGCAYPF GCTWGCAYPF AGCTWGCAYPF AGCTWGCAYPF (underlined residue released at each cycle) 2 75% Gly(G) 25% Ala(A) GCTWGCAYPF S C N O R1 8 H2N N (N) amino acids R2 O PITC S N R1 O N O PTC polypeptide R2 O N N S 50% Cys(C) 50% Gly(G) R1 N H O H2N (N-1) amino acids R2 Specific PTH-amino acid Biochemistry Lecture 6 January 24, 2013 Fragmentation: Because it is not possible to sequence proteins larger than ~75 residues it is necessary to fragment the protein to extend the sequence information. After cleavage, the individual peptide fragments are separated from each other and each is subject to N-terminal sequencing using the Edman degradation method. a. Cyanogen bromide (CNBr) cleaves the peptide bond after Methionine residues. You do not need to know the mechanism, just the specificity of the reaction. CNBr Ser Met Gly Ala Phe Arg Leu Ile Ser Met Gly Ala Phe Arg Leu Ile b. Chymotrypsin hydrolyzes the peptide bonds that follow large hydrophobic residues, e.g. Phenylalanine, Tyrosine, Tryptophan. You should remember this cleavage pattern. Chymotrypsin Ser Met Gly Ala Phe Arg Leu Ile Ser Met Gly Ala Phe Arg Leu Ile c. Trypsin hydrolyzes the peptide bonds that follow positively charged residues, e.g. Lysine and Arginine. You should remember this cleavage pattern as well. Trypsin Ser Met Gly Ala Phe Arg Leu Ile Ser Met Gly Ala Phe Arg Leu Ile Sequence Strategies: If only two fragments are produced by the cleavage reaction, then it is straightforward to reconstruct the sequence using the known amino terminal sequence of the original protein to determine which fragment is first. i) Obtain sequence past cleavage point (X) ii) Cleave, purify fragments, sequence 2nd fragment. However if the original protein is cleaved into three or more fragments, or they cannot be sequenced in their entire length, then it is not possible to determine the correct order of fragments using a single cleavage agent. Multiple overlapping fragments have to be used to determine the correct ordering: K frag. A K Y Y K Y Y K frag. B Y Y K K Y K frag. D Y Y Y frag. E frag. D Y K frag. F K Y frag. C Y Y frag. E overlapping sequences 3 Original protein frag. B K Cleavage with trypsin [Lys(K)/Arg(R)] Two possible solutions using trypsin fragments (assuming amino-terminal seq. is known). K K Y frag. A frag. C K Y K Original protein K K frag. F Cleavage with chymotrypsin [Tyr(Y)/Phe(F)/Trp(W)] Overlapping sequences from second cleavage reagent indicates correct order of trypsin fragments. Biochemistry Lecture 6 January 24, 2013 An example: Ala-Gly-Met-Ser-Thr-Gly-Val-Val-Lys-Gly-Ser-Ala-Phe-Leu Ala Gly Met Ser Thr Gly Val Val Lys Gly Ser Ala Phe Leu Here I have assumed that 7 cycles of Edman degradation are possible; note ~75 are more typical. A. The first 7 cycles of Edman sequencing on the intact peptide released the following amino acids, in this order: Ala, Gly, Met, Ser, Thr, Gly, Val, therefore , the initial, amino-terminal sequence of the peptide is: Ala Gly Met Ser Thr Gly Val Val Lys Gly Ser Ala Phe Leu B. A new sample of the intact peptide was treated with CNBr. The two peptides (CNBr-1, CNBr-2) that were produced were isolated and each was subject to sequencing, giving the following sequences (The last three residues (Ala-Phe-Leu )could not be determined due to limitation in the Edman degradation to 7 steps, these are highlighted gray). Ala Ala Gly Gly Met Ser Thr Met Gly Ser Thr Val Val Lys Gly Val Val Gly Lys Ser Ala Gly Phe Ser Ala Leu Phe Leu C. A new sample of the peptide was treated with Trypsin. The two peptides (Trp1, Trp2) that were produced were isolated and each was sequenced. Al a Al a Gl y Gl y Met Met Ser Ser Thr Thr Gl y Gl y Val Val Val Val Lys Gl y Ser Gl y Lys Al a Ser Phe Al a Leu Phe Sequence alignment strategy: Find overlaps between fragments obtained with different cleavage reagents & use these overlaps to correctly order the peptides obtained from one sequencing reaction. The overlaps can be readily identified by finding a cleavage site in a peptide that would be cut by another cleavage reagent and then identifying the correct fragment based on the expected amino-terminal sequence. A) The original amino-terminal sequence contains a Methionine (Met), so I’d look for a CNBr fragment beginning with Ser-Thr-Gly-Val, the information from that CNBr fragment extends the sequence: Ala Gly Met Ser Thr Gly Val Ser Thr Gly Val Val Lys Gly Ser Ala Phe Leu B) The lysine indicates that there should be a trypsin fragment beginning with Gly, the sequence of that trypsin fragment allows the completion of the sequence: Ala 4 Gly Met Ser Thr Gly Val Ser Thr Gly Val Val Lys Gly Ser Ala Phe Leu Gly Ser Ala Phe Leu Leu