CH1: localized chemical bonds
advertisement

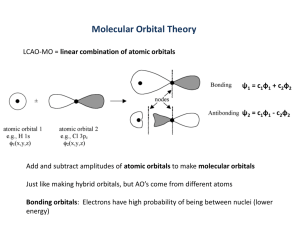
CHEM 73/8311 cs1-bonding-structure.docx 2016Jan20 1 Localized chemical bonds 1 bonds are the net interaction of electrons with nuclei (Traylor and Traylor) 1.1 energy determined by position in space relative to nuclei and electrons 1.2 electrons occupy molecular orbitals, MO 1.3 MO = linear combination of atomic orbitals 2 MO properties based on atomic orbital properties 2.1 if AO’s have high energy then MO’s have high energy 2.2 energy related to distance from nuclei and number of nodes (i.e. where = 0) 2.3 Compare the wave functions for 1s, 2p, and 2s orbitals in Figure 1-5.bmp 2.3.1 Two components radial (r) and angular () 2.3.2 1s Ke Zr a0 Zr Zr Zr Zr 2 pz K e 2 a0 cos 2 s K 2 e 2 a0 a0 a0 K =constant, r = distance from nucleus, a0 = atomic radius, = angle with Z axis 2.3.3 All orbitals have a node at r = , p obital has a node at r = 0 2.3.4 2p orbital also has angular dependence, = 0 at 90 and 270 o 2.3.5 2s orbital has a node at 2 - Zr/ao i. e. changes sign 2.3.6 3s has two nodes, as does 3p, what about 3d? 2.3.7 # of finite nodes = quantum level - 1 2.4 nodes are apparent on macroscopic level: guitar string and waves (Figure 1-3.bmp) 2.5 angular properties are shown in figure 1-10.bmp 2.6 figure 1-9.bmp shows nodal properties 2.7 Signficance of sign of ? No physical significance, determines the overlap of orbitals consider s and p, or p and d overlap 2.8 Electron density is related to (r): figure 1-6.bmp 2.8.1 total density at distance r from nucleus is 4r2(r): figure 1-6.bmp 2.8.2 note that density goes up near nucleus but the volume (4r2dr) goes down 2.9 The size of the orbital changes as the effective nuclear charge decreases: see 2s and 2 p orbitals, Figure 1-16.bmp 3 atomic orbitals added and substracted to make molecular orbitals 3.1 natural concept: electrons occupy space similar as in atoms, orbitals perturbed by other nuclei 3.2 MO for two atoms and two orbitals c A A cB B Figure 2-2.bmp 3.3 Plot the mathematical expression: Figure 2-3.bmp 3.3.1 Note two components of are identical but offset by half the bond length! 3.4 Number of orbitals does not change #AO’s = # MO’s CHEM 73/8311 cs1-bonding-structure.docx 2016Jan20 3.5 Form bonding and and antibonding MO’s c A A cB B , 2 3.6 and *, formed from overlap of orbitals with no node along bond axis 3.7 and * formed from overlap of orbitals with one node along bond axis 3.8 overlap is maximized versus charge repulsion 3.9 Only valence orbitals have significant overlap: Figure 2-4.bmp 4 3.10 p-p and d-p overlap, Figure 2-8.bmp 3.11 orbitals only effectively overlap with correct symmetry along bond axis Hybridization 4.1 mathematically similar to bond formation 4.2 superposition of orbitals on one nucleus, Figure 3-7.bmp 4.3 advantage of hybridization? directional: Figure 3-9.bmp 4.4 Plot of overlap integral = S = 0 12d 4.4.1 Note s-s () and p-p () orbitals has a unit overlap at r = 0 4.4.2 s-p overlap is zero (not shown) at r = 0 4.4.3 p-p () has small overlap and goes to zero at r = 0 4.4.4 Note that sp, sp2, and sp3 have large overlap at bonding distances (~1 Å) 4.4.5 sp, sp2, and sp3 “point” in one direction unlike s or p. 4.5 How do you determine hybridization? 4.5.1 conservation of obitals: count bonds, one p used for each bond 4.5.2 remaining p and s orbitals are hybridized. 4.6 sp3 hydridization implies 4 identical orbitals methane 4.6.1 PES: photoelectron spectroscopy 4.6.2 Consider PES for boron atom: it should have 3 bands, 1s2, 2s2, 2p (2 valence bands) 4.6.3 CH in methane, apparently has 4 identical bonds 4.6.4 methane should have one valence band for 4 bonds and one band for the carbon 1s2 4.6.5 PES of methane indicates there are not 4 bonds using 4 equivalent sp3 4.6.6 PES gives two valence bands, 14 (6e in t2 level, 3C 2p and 4H 1s) and 23 ev (2e in a1 level, carbon 2s and 4H 1s) a1 t2 4.6.7 Differences not detectable by other techniques CHEM 73/8311 cs1-bonding-structure.docx 2016Jan20 3 Physical/Chemical Properties 1 electronegativity - tendency to attract electron density, determines reactivity of molecule 2 dipole - created in molecule by differences in electronegativities and/or electron delocalization (azulene), center of negative and positive charges at different positions, dipole increases with distance between centers and magnitude of charges 3 inductive effects - atoms or functional groups polarize adjacent groups (attraction or repulsion of charge) through bonds (polarized bonds create electron deficiency or electron excess 4 field effects - polarization through space (example of one independent of the other? diammonium norbornane?) 5 bond lengths - constant unless atoms change, orbitals change, substituents change (show examples) 6 bond angles - varies with hybridization, functional groups (electronegativity and steric bulk), lone pairs 7 p orbital better donor than s (why?), electronegative group bonds with orbitals of greatest p character, remaining orbitals have more s character (i. e., CH3Cl) Bond Strengths show examples 1 stronger with better overlap and low energy atomic orbitals, CC versus CO 2 strong bonds with electronegative atoms, H (good overlap without repulsion) http://www.cem.msu.edu/~reusch/OrgPage/bndenrgy.htm#dissbe http://www.sartep.com/chem/chartsandtools/bondenergy.cfm 3 shorter onds are stronger, more s character, bonds are weaker with steric repulsion or conjugation, stability of fragments 4 bond dissociation energy - specific to molecule and site like norbornane v heptane 5 average bond energy - average from several compounds 6 bond energies can be used to calculate heats of reaction = difference of bonds formed and broken: methane and chlorine, HCl (102 kcal/mol) ClCl (57) 7 affect how fast bonds are broken and equilibria Delocalized chemical bonds (electrons) 1 evidence of resonance energy 1.1 definition: extra energy associated with alternate Lewis structures 1.2 Often resonance energy is overestimated 1.2.1 calculated enthalpy of formation from bond energies based on non-resonant compounds 1.2.2 does not take into account stabilization due to hybridization changes CHEM 73/8311 cs1-bonding-structure.docx 2016Jan20 4 1.2.3 benzene versus cyclohexatriene 1.2.4 cyclohexene H(hydrogenation) = -28.6 kcal/mol, benzene = -49.8 kcal/mol ≠ 3x(-28.6) = -85.6. sp3- sp2 C-C bond not a good model for cyclohexatriene sp2-sp2single bond 2 1.3 equivalent bond lengths in benzene definitive evidence of resonance 1.4 butadiene is planar, barrier is not steric effect 2 HMO: Huckel Molecular Orbitals 2.1 orbitals are formed from adjacent p orbitals of conjugated polyenes and related compounds 3 2.2 Signs/nodes pattern same as guitar string 2.3 Number of nodes increase as energy increases 2.4 Alternating plane and C2 symmetry (signs and coefficients: latter changes with diffent nuclei) 2.5 Relative energies can be calculate from simple formula j 2.5.1 E m j , m j 2 cos for j = 1 to n (all carbon) n 1 4 2.5.2 E is the energy of a molecular orbital with one electron 2.5.3 n is number of carbon atoms in chain 2.5.4 j indicates specific molecular orbital 2.5.5 is Coulomb integral, energy of p orbital (negative) 2.5.6 is resonance integral (negative) bonding energy of 1e in ethane orbital 2.5.7 n = 2 for ethene, 2cos/3 = 1 for j= 1, therefore is bonding energy in ethane 2.5.8 enegies of orbitals are symmetrical above and below 2.5.9 odd polyenes have extra orbital where E = when j/n+1 = ½ 2.5.10 middle orbital always has same energy as p orbital 2.6 HMO obtained by mixing atomic orbitals: c11 c 22 c 33 ... 2.6.1 Coefficients determine how much of each atomic orbital 2.6.2 Coefficients are normalized and total contribution = 1 c12 c 22 c 32 ... 2.6.3 exact calculation coefficients of MO, in text 2.7 examine MO's for ethene, allyl, butadiene, pentadiene 2.7.1 node occurs when sign of adjacent orbitals change 2.7.2 First HMO has no node 2.7.3 Each subsequent HMO has additional node 2.7.4 Atomic orbitals with same sign have bonding interaction 2.7.5 orbitals with opposite sign have antibonding interaction CHEM 73/8311 cs1-bonding-structure.docx 2016Jan20 5 2.7.6 consider empty, radical, lone pair conjugation in allylic system 2.7.7 butadiene electrons are lower and higher in energy then ethene's 2.7.8 compare reactivities and energies of methyl anion, ethene and allyl anion, (similarly for cation, ethene and allyl cation) 3 CH2=CHCH2 perturbation theory CH2=CHNH2 3.1 allyl anion v enamine v carboxylate 3.2 cross conjugation – ketone versus ester pKa competing conjugation inhibited 4 favored resonance forms 4.1 compare Lewis structures energetically 4.2 electrons move but atoms do not 4.3 maximize number bonding electrons (ketone versus amide) 4.4 atoms contributing orbitals to delocalized bonds are coplanar 4.5 forms with charge separation and fewer bonds are higher energy 4.6 no distorted bond angles or length (norbornanone, amide at bridge) 4.7 negative charge is on most electronegative atoms, etc. (nitrosomethane) 5 p-d bonding and ylids O H3C S O CH3 H3C S R R R P R CH3 CH2 R P CH2 R 5.1 bonds with S and P with neutral formal charge have dipolar resonance form 5.2 p-dbonds are ussually to O and C 5.3 for C, known as ylide, signifcant charge on C makes it good nucleophile 6 Hyperconjugation 6.1 no-bond resonance form 6.2 a better picture: + MO, 3-center two-electron bond H H H H H H 2 p H H -CH H H H H H H, H H H H H H , Tautomers 1 structural isomers in rapid equilibrium, usually proton shifts 1 CHEM 73/8311 cs1-bonding-structure.docx 2 2016Jan20 6 keto-enol, most enols are unfavorable, can be important intermediates, favored by stabilization of OH or C=C OH OH OH OH Ph OH H Ph 9.1% 6x10-7% 4x10-5% O O H2C O H O OEt none O Ph OH H3C 80% O 100% O O OEt Ph 3 nitroso-oxime, CH3-N=O CH2=NOH 4 nitro-aci, CH3-NO2 CH2=NO2H 5 imine-enamine, CH3CH=NH CH2=CHNH2 Non-covalent bonding 1 hydrogen bonds: three-center 4 electron bonds 1.1 AH-B, three center MO, compare with two center 1.2 A and B = O, N or F 1.3 most are between 3-6 kcal/mol 1.4 lifetime on order of picoseconds 1.5 linear, repulsion of A- and B1.6 detection of H bonding: AH stretch, lower hydrogen bonding frequency with 1.7 CH hydrogen bonding, for ethyne, hydrogen cyanide, chloroform (related halocarbons) 1.8 SH forms weak hydrogen bonds, S and P can be lone-pair donor 1.9 F-> Cl-> Br-> I-, carbanions and isonitriles can be lone pair donors 1.10 2 2.1 very weak hydrogen bonds with electrons and cyclopropane electron donor-acceptor complexes coordination complexes, donation and backbonding, get for benzene, http://instruct.uwo.ca/chemistry/373f/Nifty%20Stuff/aromaticity%201.htm 2.2 olefin-Ag+, C6H6Cr(CO)3 H OCH2CH3 CHEM 73/8311 cs1-bonding-structure.docx 2016Jan20 Ag Ag Ag O 3 7 Cr C C C O O Cr Cr charge transfer complexes: ionic bonds 3.1 electron transfer between highly oxidized (aromatics/olefins with electron withdrawing groups) and reduced (amines and olefins with amine and hydroxyl groups) compounds NH2 O N N C C C C N NH NH2 4 O N N HN N cryptands and related compounds 4.1 poly hetero cyclic and polycyclic compounds form complexes with ions, size selective, usually ion-dipole interaction