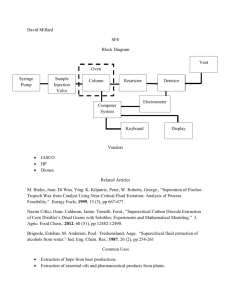
Extending the molecular size in accurate quantum - IFF-CSIC

CODECS 2013 Workshop. San Lorenzo de El Escorial, Madrid, 18
th
–22
nd
April, 2013
Extending the molecular size in accurate quantum-chemical calculations: the equilibrium structure and spectroscopic properties of biomolecules
Cristina Puzzarini a , Malgorzata Biczysko b , Julien Bloino c,d , Vincenzo Barone d a Dipartimento di Chimica “Giacomo Ciamician”, Universitá di Bologna, Via F. Selmi 2, I-40126
Bologna, Italy b Center for Nanotechnology Innovation @NEST, Istituto Italiano di Tecnologia, Piazza San
Silvestro 12, I-56127 Pisa, Italy c Consiglio Nazionale delle Ricerche, Istituto di Chimica dei Composti OrganoMetallici
(ICCOM-CNR), UOS di Pisa, Area della Ricerca CNR, Via G. Moruzzi 1, I-56124 Pisa, Italy d Scuola Normale Superiore, piazza dei Cavalieri 7, I-56126 Pisa, Italy cristina.puzzarini@unibo.it
From the experimental point of view, there is an increasing consensus that a detailed knowledge of the behavior of the main building blocks of biomolecules (amino acids, nucleic bases and carbohydrates, etc.) without the perturbing effect of environment (unavoidable in condensed phases) is a mandatory prerequisite toward the understanding of the role played by different interactions in determining the biological activity in terms of structure-activity relationships. This has stimulated our investigation toward the set up of an accurate computational strategy, which is still affordable for biomolecules. A state-of-the-art quantumchemical approach for the accurate evaluation of molecular structures as well as spectroscopic properties is presented and validated on the basis of accurate experimental data. The integrated theoretical model proposed is based on accurate post-Hartree-Fock computations (involving composite schemes) of energies, structures and properties coupled to DFT corrections for the proper inclusion of vibrational effects at an anharmonic level (as provided by second-order perturbation theory).
In this frame, the equilibrium structures of glycine, uracil and thiouracil have been determined by means of composite schemes that rely on extrapolation techniques (based on hierarchic basis set families) and the proper inclusion of electron-correlation effects, 1-4 whereas the integrated approaches mentioned above allowed us to derive semi-experimental equilibrium geometries and rotational parameters. While in the case of glycine and uracil the available experimental data have been used to verify the accuracy of our computed results, the experimental investigation of the rotational spectra of thiouracil and of the thiouracil-water complex has been guided by our calculated spectroscopic parameters.
Acknowledgments
This work was supported by Italian MIUR (PRIN 2009) and by the University of Bologna (RFO funds). The support of COST-CMTS Action CM1002 ``COnvergent Distributed Environment for
Computational Spectroscopy (CODECS)'' is also acknowledged..
References
1.
M. Heckert, M. Kállay, J. Gauss, Mol. Phys., 103 , 2109 (2005).
2.
M. Heckert, M. Kállay, D. P. Tew, W. Klopper, J. Gauss, J. Chem. Phys., 125 , 044108 (2006).
3.
C. Puzzarini, V. Barone, Phys. Chem. Chem. Phys., 13, 7158 (2011)
4.
V. Barone, M. Biczysko, J. Bloino, C. Puzzarini, J. Chem. Theo. Comput., in press
CODECS 2013 Workshop. San Lorenzo de El Escorial, Madrid, 18
th
–22
nd
April, 2013
(2013), doi:10.1021/ct3010672