Weakness and Hypotonia
Dr. William W. C. Young
Pediatric Neurologist
Unending List of Causes of Weakness
Duchenne
muscular
dystrophy
Becker’s
muscular
dystrophy
Pompe’s disease
McArdle’s
disease
Carnitine
palmitoyl
transferase
deficiency
Medium chain
acyl-coA
DHG def
Cushing’s
disease
Nemaline rod
myopathy
Dermato
myositis
Botulism
Myasthenia
gravis
Organophospha
toxicity
Magnesium
toxicity
Eaton Lambert
syndrome
Endplate AchR
deficiency
Endplate AchE
deficiency
Choline acetyl
transferase
deficiency
Sea snake
venom
Gentamycin
toxicity
Viper venom
Guillain Barre
syndrome
Diphtheria
Vincristine
neuropthy
Diabetic
neuropthy
Polyarteritis
nodosa
Metachromatic
leuko
dystrophy
Cockayne
syndrome
Charcot Marie
Tooth
disease
Refsum’s
disease
Vitamin E
deficiency
Spinal muscular
atrophy
Poliomyelitis
Werdnig Hoffman
disease
Kugelberg
Welander
disease
Cold exposure
Radiation sickness
Fabry’s disease
Herpes zoster
Lyme disease
Hepatitis B
Porphyria
Mechanical
trauma
Acromegaly
Thallium toxicity
Arsenic toxicity
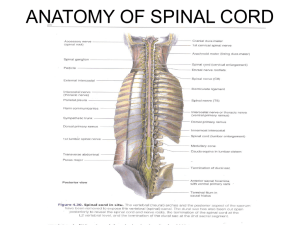
Syringomyelia
Transverse
myelitis
C1-C2 sub
luxation
Spinal cord
infarction
Vertebral injury
Spinal canal
hematoma
AV malform
Tabes dorsalis
Subacute
combine
degener
Varicella
myelopthy
Syringobulbia
Devic’s disease
Acute Dissem
EncephM
Meningitis
Hypoxic ischemic
encephalop
athy
Cerebral palsy
MELAS
MERRF
Adrenoleukodystr
ophy
Gliomatosis
cerebri
Obstructive hydro
cephalus
Status epilepticus
Substance abuse
Uremia
Liver failure
Multiple sclerosis
Hyothyroidism
Tay Sach’s disease
Systematic Neurologic Evaluation
• Identify the problem accurately
• Localize the lesion
• Derive a differential diagnosis
e.g., Abnormal Movements
• Identify the problem accurately
– Seizures?
– Dyskinesias? (tremors, tics, chorea)
– Unsteadiness?
– Stereotypic movements?
• Localize the lesion
• Derive a differential diagnosis
e.g., Abnormal Movements
• Identify the problem accurately
• Localize the lesion
– Central or peripheral or spinal cord?
– Nerve, neuromuscular junction, or muscle?
• Derive a differential diagnosis
e.g., Abnormal Movements
• Identify the problem accurately
• Localize the lesion
• Derive a differential diagnosis
– Acute (infectious, traumatic, toxic-metabolic)
e.g., Abnormal Movements
• Identify the problem accurately
• Localize the lesion
• Derive a differential diagnosis
– Acute (epileptic, infectious, traumatic, toxicmetabolic, vascular-ischemic)
– Chronic (endocrine, degenerative, neoplastic,
chronic toxicity, nutritional, autoimmune,
congenital, systemic)
Two Cases
• Two year old female that is stumbling
• 18 month old male that is not walking
Stumbling two year old female
•
•
•
•
Onset three days ago, unchanged
Recent respiratory infection 4 weeks ago
Prior history unremarkable
No toxic exposure, no recent travel, no
adventure in the woods, no med use
• No history of trauma
• Normal developmental milestones
Stumbling two year old female
•
•
•
•
•
•
•
Speaking in short phrases
No dysmorphic features, normal head circ
Normal cranial nerves
No nystagmus, no unsteadiness
No abnormal movements
Normal reflexes
Decreased movements in left leg, pain in left
calf with squeezing
Stumbling two year old female
• Identify the problem accurately
• Localize the lesion
• Derive a differential diagnosis
Stumbling two year old female
• Identify the problem accurately
– Weakness
Stumbling two year old female
• Identify the problem accurately
– Weakness
– Pain
– Ataxia
– Vertigo
Stumbling two year old female
• Identify the problem accurately
– Weakness in left leg
– Pain in left calf
– Ataxia
– Vertigo
Stumbling two year old female
• Identify the problem accurately
– Weakness and pain in left leg
• Localize the lesion
– Central
– Spinal cord
– Neuromuscular
Stumbling two year old female
• Identify the problem accurately
– Weakness and pain in left leg
• Localize the lesion
– Central
– Spinal cord
– Neuromuscular (anterior horn cell, nerve,
neuromuscular junction, muscle?)
Stumbling two year old female
• Identify the problem accurately
– Weakness and pain in left leg
• Localize the lesion
– Central
– Spinal cord
– Neuromuscular (anterior horn cell, nerve,
neuromuscular junction, muscle?)
Stumbling two year old female
• Identify the problem accurately
– Weakness and pain in left leg
• Localize the lesion
– Neuromuscular (muscle—serum CK 2000)
• Derive a differential diagnosis
Stumbling two year old female
• Identify the problem accurately
– Weakness and pain in left leg
• Localize the lesion
– Neuromuscular (muscle—serum CK 2000)
• Derive a differential diagnosis
– Acute vs Chronic?
Stumbling two year old female
• Identify the problem accurately
– Weakness and pain in left leg
• Localize the lesion
– Neuromuscular (muscle—serum CK 2000)
• Derive a differential diagnosis
– Acute (epileptic, infectious, traumatic, toxicmetabolic, vascular-ischemic)
Stumbling two year old female
• Not Chronic
– dystrophinopathy (Duchenne, Becker’s)
– congenital myopathy (Nemaline rod,
centronuclear)
– inflammatory myopathy (dermatomyositis)
– metabolic myopathy (MELAS, MERFF, MCAD def,
CPT deficiency, Pompe’s, McArdle’s)
Stumbling two year old female
• Differential diagnosis
– anterior horn cell (poliomyelitis)
– neuropathy (Guillain Barre syndrome)
– neuromuscular junction (Myasthenic crisis,
Botulism, organophosphate poisoning)
– Muscle (trauma, inflammation, infection,
ischemia)
Stumbling two year old female
• Identify the problem accurately
– Weakness and pain in left leg
• Localize the lesion
– Neuromuscular (muscle—serum CK 2000)
• Derive a differential diagnosis
– Acute (epileptic, infectious, traumatic, toxicmetabolic, vascular-ischemic)
• POST INFECTIOUS MYOSITIS
Delayed walking 18 month male
•
•
•
•
•
No acute changes
Normal prenatal and birth history
No chronic medical problems, no meds
No hospitalizations, no surgeries
No toxic exposure, no recent travel, no
adventure in the woods, no med use
• No recent history of trauma or infection
Delayed walking 18 month male
• Normal head size, no dysmorphic features, no
neurocutaneous markers
• Normal cranial nerves
• Has 20 word vocabulary, understands verbal
• No abnormal movements or postures
• Cruising along furniture
• Can appose thumbs to radii, can dorsiflex ankles, has
vertical slip, some draping with horizont suspension
• Normal reflexes, mild head lag
Delayed walking 18 month male
• Identify the problem accurately
• Localize the lesion
• Determine the mechanism of action (to derive
a reasonable differential diagnosis
Delayed walking 18 month male
• Identify the problem accurately
– Weakness
– Pain
– Ataxia
– Vertigo
Delayed walking 18 month male
• Identify the problem accurately
– Not weak
– Not pain
– Not ataxic
– No vertigo
– Hypotonic
Delayed walking 18 month male
• Identify the problem accurately
– Weakness
– Pain
– Ataxia
– Vertigo
– Hypotonia?
– Physiologic?
Delayed walking 18 month male
• Identify the problem accurately
– Hypotonia without weakness
• Localize the lesion
– Central
– Spinal cord
– Neuromuscular
Delayed walking 18 month male
• Identify the problem accurately
– Hypotonic without weakness
• Localize the lesion
– Not central (normal verbal and social
development, normal head circ, no dysmorphic
features, no neurocutaneous markers )
Delayed walking 18 month male
• Identify the problem accurately
– Hypotonic without weakness
• Localize the lesion
– Not central (normal verbal and social
development, normal head circ, no dysmorphic
features, no neurocutaneous markers )
– Not spinal cord (no paraplegia, no weakness)
Delayed walking 18 month male
• Identify the problem accurately
– Hypotonic without weakness
• Localize the lesion
– Not central (normal verbal and social
development, normal head circ, no dysmorphic
features, no neurocutaneous markers )
– Not spinal cord (no paraplegia, no weakness)
– Neuromuscular? (no weakness, normal reflexes,
normal muscle bulk)
Delayed walking 18 month male
• Identify the problem accurately
– Hypotonia without weakness
• Localize the lesion
– Not central, not spinal cord, ?neuromuscular
•
•
•
•
Serum CK 40 normal
TSH normal
ESR 3 normal
Lactate 1.2 normal
Delayed walking 18 month male
• Identify the problem accurately
– Hypotonic without weakness
• Localize the lesion
– Not central, not spinal cord, ?neuromuscular
• Derive a differential diagnosis
– Acute vs chronic?
Delayed walking 18 month male
• Identify the problem accurately
– Hypotonic without weakness
• Localize the lesion
– Not central, not spinal cord, ?neuromuscular
• Derive a differential diagnosis
– Acute vs chronic?
Delayed walking 18 month male
• Differential diagnosis
– Muscle (dystrophinopathy, congenital myopathy,
inflammatory myopathy, metabolic myopathy)
(Duchenne, Becker’s)
– Neuromuscular junction (chronic Botulism,
myasthenia, organophosphate poisoning)
– Nerve (Guillain Barre syndrome, diphtheria,
poliomyelitis, Charcot Marie Tooth)
– Brain (genetic disorders, hypotonic cerebral palsy,
microcephaly, macrocephaly, hypothyroidism)
Delayed walking 18 month male
• Identify the problem accurately
– Hypotonic without weakness
• Localize the lesion
– Not central, not spinal cord, ?neuromuscular
• Derive a differential diagnosis
– Acute vs chronic?
Delayed walking 18 month male
• Identify the problem accurately
– Hypotonic without weakness
• Localize the lesion
– Not central, not spinal cord, ?neuromuscular
• Derive a differential diagnosis
– Acute vs chronic?
• Something distal to the muscle?
Delayed walking 18 month male
• Identify the problem accurately
– Hypotonic without weakness
• Localize the lesion
– Not central, not spinal cord, ?neuromuscular
• Derive a differential diagnosis
– Acute vs chronic?
• LIGAMENTOUS LAXITY
– Physiologic
– Ehlers Danlos syndrome, Cutis Laxa
Hypotonia (archaic terms)
• Infantile progressive spinal muscular atrophy (Wernig 1891,
Hoffman 1893)
• Myotonia congenita (Oppenheim 1900)
• Amyotonia congenita (Collier and Wilson 1908)
• Benign congenital myopathy (Batten 1903, turner 1940)
• Congenital universal muscular hypoplasia (Krabbe 1947)
• Infantile muscular atrophy (Greenfield and Stern 1927)
• Amyotonia congenita symptom complex (Brandt 1950)
• Primary (essential) hypotonia (Sobel 1926)
• Benign congenital hypotonia (Walton 1956)
Non-Neuromuscular Causes of
Hypotonia
•
•
•
•
Disorders of the central nervous system
Connective tissue disorders
Genetic disorders
Metabolic; nutritional; endocrine
Non-Neuromuscular Causes of
Hypotonia
• Disorders of the central nervous system
– Non-specific mental deficiency
– Birth trauma, intracranial hemorrhage, neonatal
hypoxic ischemic encephalopathy
– Hypotonic cerebral palsy
– Metabolic disorders; lipidoses; leukodystrophies,
mucopolysaccharidoses, aminoacidurias,
mitochondrial disorders
Non-Neuromuscular Causes of
Hypotonia
• Connective tissue disorders
–
–
–
–
–
–
–
–
Congenital laxity of ligaments
Ehlers-Danlos syndromes
Cutis laxa
Marfan syndrome
Osteogenesis imperfecta
Arachnodactyly
Loeys-Dietz syndrome
Camurati-Engelman syndrome
Ehlers Danlos syndromes
• EDS I – severe
• EDS II – mild
• EDS III – hypermobile
joints
• EDS IV – vascular
• EDS V – X linked type
• EDS VI – ocular, scoliosis
• EDS VII –arthrochalasis
multiplex congenita
• EDS VIII – periodontitis
• EDS IX – copper
transport disorder; Xlinked cutis laxa
• EDS X – fibronectin
abnormality
• EDS XI – familial
hypermobility
syndromes
Ehlers Danlos syndromes
Ehlers Danlos syndromes
Ehlers Danlos syndromes
Ehlers Danlos syndromes
Non-Neuromuscular Causes of
Hypotonia
• Genetic disorders
– Down syndrome
– Prader-Willi syndrome
– Angelman syndrome
– William syndrome
– Miller-Dieker syndrome
– Smith Lemli Opitz syndrome
– Other structural chromosomal abnormalities
Non-Neuromuscular Causes of
Hyotonia
• Metabolic; nutritional; endocrine
– Organic acidemias
– Hyperacalcemia
– Rickets
– celiac disease
– Hypothyroidism
– renal tubular acidosis
Maxims
• Most children who are weak, are hypotonic
(not all)
• Most children who are hypotonic, are not
weak
Maxims
• Most children who are weak, are hypotonic
(not all)
– exception is spastic cerebral palsy (spastic
quadriplegia, spastic diplegia)
• Most children who are hypotonic, are not
weak
– ligamentous laxity is the most common cause of
hypotonia
– Genetic conditions are not always associated with
weakness (Down syndrome, Prader Willi
syndrome, William syndrome, Smith Magenis)
Duchenne Muscular Dystrophy
• Calf pseudohypertrophy (fat
and connective tissue replaces
normal muscle)
• Positive Gower’s sign
• Genetic deficiency of muscle
protein dystrophin
• 1:3500 male births
• Large protein, with many sites
for deletions, duplications,
sequencing abnormalities
• X-linked recessive inheritance
• Serum CK over 10,000
• Xp21.2
• Possible complications
–
–
–
–
–
Cardiomyopathy
Congestive heart failure (rare)
Deformities
Heart arrhythmias (rare)
Mental impairment (varies,
usually minimal)
– Permanent, progressive
disability
• Decreased mobility
• Decreased ability to care for self
– Pneumonia or other respiratory
infections
– Respiratory failure
Guillain Barre Syndrome
• Acute inflammatory
demyelinating
polyneuropathy
• Areflexic
• Weakness, paresthesia,
limb pain
• Cytoalbuminogenic
dissociation (CSF protein
up to 1 gm after one
week)
• EMG with delayed F
waves
• IVIg or plasmaphoresis for
severe or rapidly
progressive cases
• Dysautonomia risk
• Respiratory failure risk
• Ascending paralysis
• Autoimmune disorder
Myasthenia Gravis
• Autoimmune disorder
• Thymectomy
• Paralysis from
myasthenic crisis and
cholinergic crisis
• Palliation and control
w/acetylcholinesterase
inhibitor
• AchR antibody levels
(80-90%)
• Muscle fatiguability
• Bulbar symptoms
prominent
• Different from
congenital myasthenic
syndromes
Dermatomyositis
• Heliotrope rash and Gottron’s papules
• malar erythema, poikiloderma in a
photosensitive distribution, violaceous
erythema on the extensor surfaces, and
periungual and cuticular changes.
• Perifascicular atrophy
• Calcinosis
Dermatomyositis
• Joints (arthralgia)
• Reticuloendothelial (lymphadenopathy,
splenomegaly, hepatomegaly)
• Respiratory(acute respiratory distress from lung
parenchymal involvement)
• Gastrointestinal (ulcers of stomach, intestines)
• Cardiovascular (murmurs, friction rubs, EKG changes
• Renal (albuminuria)
Pompe’s Disease
•
•
•
•
•
Autosomal recessive, 17q23
Glycogen storage disease type II
Acid maltase deficiency
Cardiomegaly and hepatomegaly
EKG with characteristic gigantic QRS
complexes and very short P-R interval
• Pseudomyotonic bursts on EMG
• Infantile form generally fatal
Charcot Marie Tooth disease
•
•
•
•
•
Hereditary sensory motor neuropathy
Champagne bottle legs
Areflexia (neuropathy)
Most common inherited neurologic disorder
CMT1A accounts for about 60% of all
autosomal dominant neuropathies, CMT2
accounts for about 22%, X-linked CharcotMarie Tooth disease (CMTX) for about 16%,
and CMT1B for approximately 1.6%.
Charcot Marie Tooth disease
• More common types
– CMT1A, CMT2 (CMT2A, CMT2B, CMT2C, CMT2D,
CMT2E, CMT2F, CMT2L, CMT2Po), CMT1B, CMTX
• Rarer types
– CMT4A, CMT4B, CMT4B2 CMT4C, CMT4D(lom),
CMT4E, CMT 4F, AR-CMT2, AR-CMT2A, AR-CMT2B
• Most associated with abnormalities of specific
gene loci
MELAS
• Mitochondrial Encephalomyopathy, Lactic
Acidosis and Stroke
• seizures, diabetes mellitus, hearing loss,
cardiac disease, short stature,
endocrinopathies, exercise intolerance, and
neuropsychiatric dysfunction
• Multisystem involvement: CNS, skeletal
muscle, eye, cardiac muscle, and, more rarely,
the GI and renal systems
MELAS
• Mitchondrial t-RNA abnormalities
• 3243 A → G mutation (80% of cases) produces
a severe combined respiratory chain defect in
myoblasts, with almost complete lack of
assembly of complex I, IV, and V, and a slight
decrease of assembled complex III.
• Altered mental status, schizophrenia, bipolar
symptoms, autistic spectrum disorder
MERRF
•
•
•
•
Myoclonic Epilepsy with Ragged Red Fibers
Mitochondrial encephalomyopathy
Ataxia, lactic acidosis
Less often: dysarthria, optic atrophy, short stature,
hearing loss, dementia, and nystagmus
• Progressive multisystem disorder
• Mitochondrial DNA , mutation in A8344G most
common, T8356C less often
MERRF
Normal muscle (H & E)
Ragged Red Fibers (Gomori Trichrome)
McArdle’s disease
•
•
•
•
•
Autosomal recessive, 11q13
Glycogen storage disease Type V
Muscle phosphorylase deficiency
Cramps on exertion, sometimes myoglobinuria
Easy fatiguability in childhood and
adolescence, diagnosis usually in adulthood
• No cardiac involvement
Botulism
•
•
•
•
•
•
Dysphagia, ptosis
Fixed dilated pupils
Neuromuscular junction blockade
Clostridium botulinum toxin A
Intestinal colonization under 12 months of age
Diplopia, dysarthria, dry mouth, sore throat,
dysphonia, nystagmus, ataxia, paresthesia
• Paralytic ileus, constipation, urinary retention, poor
anal sphincter tone
MCAD deficiency
• Medium chain acyl-coA DHG, chrom 1p31
• Fatty acid metabolism difficulty
• Exaggerated lethargy accompanied by
vomiting and acidosis with previous viral
illness, quick recovery with IV fluids
• No ketones on urinalysis
• increased preprandial irritability, lethargy,
jitteriness, sweating, and, possibly, seizures,
which are all symptomatic of hypoglycemia.
Spinal Muscular Atrophy
•
•
•
•
•
•
•
Werdnig Hoffman (type 1), onset 2-3 months
Kugelberg Welander (type 3)
Autosomal recessive, 5q11-q13
Tongue fasciculations, areflexia, dysphagia
Facial muscles spared
Diaphragmatic breathing
Mild arthrogryposis
ADEM
• Somewhat different than Multiple Sclerosis, 10% do progress to Multiple
Sclerosis
• Postinfectious from Herpes simplex, HHV6, CMV, EBV, varicella,
Mycoplasma
• Immunizations for rabies, pertussis, measles, mumps, tetanus, influenza
• White matter demyelination at nexus with cortical gray matter
• 1.5% mortality, seizures at onset in 25%
• Treatment with IVIg, IV cyclosporin, high dose steroids, plasmapharesis
• Emotional lability (other psychopathology) leads to limbic encephalitis
• Cannot use CSF myelin basic protein and IgG index to separate from MS
Poliomyelitis
• Poliomyelitis is an enteroviral infection that can manifest in 4 different
forms: inapparent infection (90-95%), inapparent abortive disease (510%), nonparalytic poliomyelitis (5%), and paralytic poliomyelitis (5%)
• Poliovirus is an RNA virus that is transmitted through the oral-fecal route
or by ingestion of contaminated water. Three serotypes are able to cause
human infection. The incubation period for poliovirus is 5-35 days. The
viral particles initially replicate in the nasopharynx and GI tract and then
invade lymphoid tissues, with subsequent hematologic spread. After a
period of viremia, the virus becomes neurotropic and produces
destruction of the motor neurons in the anterior horn and brainstem. The
destruction of motor neurons leads to the development of flaccid
paralysis, which may be bulbar or spinal in distribution.
Poliomyelitis
• A 4-fold increase in the immunoglobulin G (IgG) antibody titers or a
positive anti-immunoglobulin M (IgM) titer during the acute stage is
diagnostic.
• Patients who have recovered from poliomyelitis occasionally develop a
postpoliomyelitis syndrome, in which recurrences of weakness or fatigue
are observed and which usually involve groups of muscles that were
initially affected. This postpolio syndrome may develop 20-40 years after
infection with poliovirus.
Transverse Myelitis
•
•
•
•
•
•
•
•
•
•
More common, older than 5 years of age
Initial discomfort and pain, with weakness
80% thoracic, 10% cervical
Some with fever and meningismus
Paraplegia, sensory loss, sphincter dysfunction, bilateral weakness,
sensory level
MRI shows T2 increased signal in affected level of spinal cord, swelling
CSF pleocytosis in 25%, increased CSF protein in 50%
Steroids, IVIg, plasmapharesis do not help
80-90% recovery, 50% with excellent recovery
Consider Devic’s disease when accompanied with optic neuritis