March 29, 2013 - Hassanian Toma
advertisement

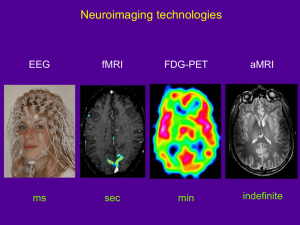
Neurology Case of the Week HASSANAIN TOMA, MD NEUROLOGY PGY-4 M A R C H 2 9 TH, 2 0 1 3 Chief Complaint Difficulty balancing when walking. HPI - Acute unsteady in gait tendency to fall – x6 weeks. - Difficulty with puzzles and going up/down stairs. - Progressively worsened over past 2 weeks . - Deny- mental status change, posturing, eye deviation, impairment of responsiveness, irritability, nausea, vomiting. PAST MEDICAL/ SURGICAL/ BIRTH HISTORY: born full term by vaginal delivery. Uncomplicated pregnancy, labor and delivery. received routine newborn care and then went home with mother. Adverse Reactions/Allergies: FAMILY HISTORY: GROWTH/DEVELOPMENT: Was normal and appropriate for the age Word sentences, uses about 50 words Deny neurologic or neurodevelopmental issues. SOCIAL HISTORY: MEDICATIONS: Milk Products(Rashes) ibuprofen PRN Lives at home with 24 yo mother (healthy) & and maternal grandparents Father is 21 yo, healthy Goes to daycare 3 days a week as mother is still in college IMMUNIZATIONS: Up to date Physical Exam General: Awake, alert, appears of stated age, active boy. Head/Neck: normocephalic. No neck masses. Eyes: PERRL. Conjunctiva clear. Fundi benign. ENT: TM's pearly and nonbulging bilaterally. No erythema or exudate of oropharynx. Dry lips. Chest: CTAB, no wheezing. CV: RRR, no murmurs, rubs, or gallops. Abdomen: abdomen is soft. Non distended. +BS Lymph: No cervical LAD. Skin: No rashes seen on visible skin. Mental State: Awake, alert, oriented to self. Cooperative and follows simple commands. Speech: clear, non-dysarthric. CN II: PERRL, visual fields full to confrontation. CN III, IV, VI:EMOI, no nystagmus. CN V: sensation intact. Strong jaw clench. CN VII: Symmetrical facial movement. CN VIII: grossly intact. CN IX & X: Gag present, uvula midline. CN XI: strong shoulder shrug. CN XII: tongue midline. Motor: Symmetrical strength, appropriate for age U/E. Weakness b/l LE with decreased tone. Helps with arm when getting up from supine. Sensory: intact to light touch, pin prick. Reflexes: 2 b/l UE, 3 b/l LE. 2 beats clonus b/l. downgoing toes. Coordination: Mild ataxia b/l Gait: wide-based, unsteady, ataxic pattern. Labs HEMATOLOGY WBC HGB HCT Platelet Normal diff 12.3 12.1 35.5% 294 SED rate 8 CHEMISTRY Sodium Potassium Chloride Carbon Dioxide Anion Gap Calcium Glucose BUN Creatinine 141 4.2 104 24 13 10.4 91 9 0.23 Protein Total Albumin Bilirubin, Total Bilirubin, Direct Bilirubin, Indirect AST ALT 7.4 4.8 0.7 0.0 0.6 41 29 Alk Phos 215 What?? Where?? Vegetative, Autonomic, Endocrine Disorders Convulsion/grand mal seizure Post-ictal status Hydrocephalus Syndrome Reference to Organ System Ataxia, cerebellar, acute Cerebellar disorder Cerebellar syndrome, acquired Heirarchical Major Groups Brain stem disorders Drugs Anticonvulsants Administration/Toxicity Benzodiazepines Administration/Toxicity Drug reaction/Side effect Tricyclic antidepressant Administration/Toxicity Antihistamine Administration/Toxicity/effect Benzodiazepine toxicity/overdose Tricyclic overdose Carbamazepine (Tegretol/Carbatrol) adm Phenytoin (Dilantin) Administration/Toxicity Poisoning (Specific Agent) Poisoning Solvent/inhalation/Huffing propane etc Glue sniffing/ingestion Gasoline poisoning Trauma Causes Concussion Head trauma Arteriosclerotic, Vascular, Venous Disorders Cerebellar hemorrhage/hematoma Infectious Disorders (Specific Agent) Viral acute illness/Viremia Labyrinthitis, viral Meningitis Bacterial Post infectious cerebellitis Infected organ, Abscesses Brain abscess Brain stem encephalitis Neoplastic Disorders Brain tumor Neuroblastoma, CNS Opsoclonic-myoclonic/paraneoplastic syn Allergic, Collagen, Auto-Immune Disorders Acute Postinfectious Cerebellitis Multiple Sclerosis Deficiency Disorders Vitamin E deficiency Congenital, Developmental Disorders Cerebral AV malformation Arteriovenous malformations Hydrocephalus, obstructive Hydrocephalus, communicating Hydrocephalus Hereditary, Familial, Genetic Disorders Ataxia-telangiectasia Myoclonic encephalopathy/childhood Cerebellar ataxia, congenital/hered. Friedreich's Ataxia MRI – T1 MRI – T2 MRI – T2 MRI – T2 MRI - FLAIR MRI – FLAIR MRI - SWI (Susceptibility) MRI – T1 w Contrast MRI – T1 w CONTRASTv MRI – T1 w CONTRASTv MRI- Differential Diagnosis Category A: severely deficient myelination predominant finding: • Pelizaeus-Merzbacher disease • Salla disease • Cockayne syndrome type II • Tay syndrome • Alpers disease • Menkes syndrome • infantile neuronal ceroid lipofuscinosis • Sandhoff disease Category B: global cerebral white matter involvement. • “infantile onset vacuolating leukoencephalopathy • “CACH” (childhood cerebellar ataxia and central hypomyelination) • “the disease of the vanishing white matter” Category C: extensive cerebral white matter abnormalities • fronto-occipital gradient and relative sparing of the occipital lobes • Alexander disease Category D: that of patients showing predominantly periventricular white matter abnormalities • Autosomal dominant leukoencephalopathy • metachromatic leukodystrophy • Krabbe disease • X-linked adrenoleukodystrophy Category E: isolated multifocal white matter abnormalities with a predominantly lobar location and relative sparing of arcuate fibers and periventricular white matter • acute disseminating encephalomyelitis • multiple sclerosis • congenital cytomegalovirus infection Category F: white matter changes in a predominantly subcortical location. preferentially involve the arcuate fibers • such as l-2-hydroxyglutaric aciduria • Kearns-Sayre syndrome Category G: white matter disorders in a predominantly posterior fossa location. • Refsum disease • cerebrotendinous xanthomatosis • adrenomyeloneuropathy MRI- Differential Diagnosis Labs Arylsulfatase A activity 59.6 nmol/hr/mg EIF Gene Sequence: Sequence analysis of the coding region of the EIF2B5 gene identified a heterozygous disease-associated missense mutation in exon 2, p.Thr91Ala (c.271A>G), missense mutation in exon 7, p.Arg339Pro (c.1016G>C) This result is consistent with a diagnosis of Leukoencephalopathy with Vanishing White Matter in this individual. Vanishing White Matter Disease NO, IT’S NOT MAGIC! What do we know? Vanishing White Matter (VWM) disease has also been known as: • Childhood ataxia with central nervous system hypomyelination • Leukoencephalopathy with vanishing white matter • Cree leukoencephalopathy - autosomal recessive neurological disease. chronic and progressive white matter disorder, exacerbated by infection or head trauma. The disease belongs to a family of conditions called the leukodystrophies. The cause of the disease are mutations in any of the 5 genes encoding subunits of the translation initiation factor EIF-2B: - EIF2B1, EIF2B2, EIF2B3, EIF2B4, or EIF2B5. It was not recognized as a clinical syndrome until the 1990s. atypical diffuse sclerosis- 1962 1st description in 1962, by Eicke et al. 36 yo F initially presented at 31 yr with gait difficulties and secondary amenorrhoea. The clinical disease course was chronic progressive with episodes of rapid deterioration after minor physical trauma. At autopsy, a diffuse, cystic destruction of the cerebral white matter was found. Around the cystic areas, a dense “network” of oligodendrocytes was seen. Only mild fibrillary astrocytosis and scant sudanophilic lipids were present. The diagnosis was “atypical diffuse sclerosis”. Epidemiology incidence and prevalence ? Perhaps one of the more common leukodystrophies. Among patients with a leukodystrophy of unknown etiology 40% met clinical criteria for VWM, 23% met MRI criteria At the time of these studies, genes were not defined. Unaffected Cree adults population: 1/10 adults were heterozygous for a G584A mutation on the EIF2B5 gene suggesting a high carrier rate in select populations Prevalence is only limited by the physician’s ability to identify the disease. As of 2006, more than 200 people have been identified with VWM, many of whom were originally diagnosed with an unclassified leukodystrophy. Clinical Presentation Febrile illness, minor head trauma, or severe fright often leads to sudden neurologic deterioration prominent ataxia and spasticity, optic atrophy and seizures can occur. Loss of motor functions, irritability, vomiting, coma, and even fever. coma, with incomplete recovery . Intellectual functioning is usually relatively spared. There are now five clinical subtypes: 1. Antenatal- 3rd trimester with decreased fetal movements & oligohydramnios. 2. Infantile- 1st yr of life - severe type 3. Early childhood- 1-5 yrs of age- most common variant, 4. Late childhood/juvenile- 1 - 5 yrs of age. 5. Adult- over 15 yrs - same as childhood and juvenile but spasticity > ataxia Genetics 5 genes that encode subunits of the eukaryotic translation initiation factor eIF2B. The eIF2B protein complex is composed of 5 different subunits eIF2Balpha, eIF2Bbeta, eIF2Bgamma, eIF2Bdelta, and eIF2Bepsilon. The genes are named EIF2B1, EIF2B2, EIF2B3, EIF2B4, and EIF2B5 located on chromosomes 12q24.3, 14q24, 1p34.1, 2p23.3, and 3q27, respectively. Genotype-phenotype correlations Phenotypic heterogeneity among members of the same family and among individuals with the same mutation phenotype is influenced by the environment and other genetic factors? However, certain genotype-phenotype correlations have been described, as illustrated by the following examples Pathogenesis initiation factors needed for translation of mRNA to polypeptides. The eIF2B protein complex initiation of protein synthesis and the cellular response to stress by activating the initiation factor eIF2. During periods of stress- trauma, heat, and infection. causes the misfolding of proteins. decreased activity of eIF2B disrupts the cellular response to stress. glial cells have a selective vulnerability to decreased eIF2B activity. Investigations Blood tests: Various abnormal findings Elevation of serum aminotransferases and lactic dehydrogenase indicates liver dysfunction Elevation of creatine kinase, urea nitrogen and amylase indicates concomitant involvement of the muscles, kidneys and pancreas respectively. Genetic Testing Genetic testing — Approximately 90% of individuals diagnosed by clinical and MRI criteria have mutations in one of the five causative. More than 120 mutations of EIF2B genes have been reported. Mutations in the EIF2B5 gene account for more than one-half of cases . Genetic testing may be particularly important to establish the diagnosis of VWM in patients with neonatal or early infantile-onset variants, as brain MRI features in early-onset disease may be atypical and are unlikely to meet MRI criteria for VWM. Labs Laboratory testing — Routine laboratory tests are generally normal in patients with VWM. Standard cerebrospinal fluid (CSF) analysis is likewise normal. A reduced CSF asialotransferrin/total transferrin ratio <8% sensitivity & specificity of 100 and 94 percent, respectively, for the identification of patients with eIF2B mutations. Because the test is inexpensive and rapid (<48 hours), the investigators suggested that determination of asialotransferrin/transferrin ratio could be used to screen cases with a suspected diagnosis of VWM prior to genetic mutation analysis, which can take weeks to months before results are available. However, the data set included no patients with neonatal or infantile onset, and further confirmation is needed to determine if the test is useful in these subgroups. Currently, determination of the asialotransferrin/transferrin ratio by two- dimensional gel electrophoresis is available only on a research basis. MRI- Dx Criteria hyposignal on T1-weighted sequences imaging (A, D) hypersignal on T2 (B, E, G FLAIR sequences imaging (C, F, H, I), infantile (C; arrow) childhood (F; arrow) juvenile/adult onset form these areas are restricted to the frontal white matter on FLAIR sequences (H; arrow). Pathology A coronal section of the left cerebral hemisphere shows the rarefaction of the white matter with microcystic changes in the periventricular area. The U-fibers, corpus callosum, and internal capsule are relatively spared (anti-MBP stain). At autopsy, gross sections of the brain demonstrate extensive cystic, gelatinous, or cavitary degeneration involving the periventricular and immediate subcortical white matter, with partial sparing of the U-fibers. Symmetric brainstem lesions within the central tegmental tracts may be present. Microscopically, the white matter demonstrates marked rarefaction, severe loss of myelin with spongy degeneration, vacuolation, astrocytic gliosis, and macrophage proliferation in some areas. Myelin loss is seen in the retrobulbar optic nerve in some patients Treatment Prognosis is often grave There is no cure or specific treatment for VWM. Avoidance of stressful situations is crucial to the management, contact sports should be avoided. Since infection and fever can have deleterious effects, antibiotics and antipyretics should be used liberally vaccinations, such as the influenza vaccine, should be kept up-to-date. Physical therapy, rehabilitation, and antispasmodic medications can be used to improve the gait and tone. Antiepileptic drugs are used in patients with epilepsy. Melatonin has been shown to provide cytoprotective traits to glial cells exposed to stressors such as excitotoxicity and oxidative stress. OUR PATIENT- 4 month f/u NEUROLOGIC EXAM: MOTOR: slightly increased tone in his legs greater than his arms. REFLEXES: 3 throughout , upgoing toes b/l, sustained ankle clonus b/l. COORDINATION: subtle ataxia noted on reaching for objects. MOBILITY: dramatically regressed in his ability to walk since his initial diagnosis nonambulatory, significant truncal ataxia on sitting. He can still crawl and stand with help followed by Rehabilitation for tone management and equipment needs He does have AFOs in place and does have a walker which they are preparing to start using They are doing daily stretches on him and he is getting PT and OT once every other week Mother avoids head trauma, and infections, fever Goes to daycare 3 days a week, on prophylactic amoxicillin daily. genetic counseling given – mother not interested. References 1. Fogli A, Wong K, Eymard-Pierre E, et al. Cree leukoencephalopathy and CACH/VWM disease are allelic at the EIF2B5 locus. Ann Neurol 2002; 52:506. 2. Eicke WJ. Polycystische umwandlung des marklagers mit progredientem verlauf. Atypische diffuse sklerose? Arch Psychiat Nervenkr 1962; 203:599. 3. van der Knaap MS, Pronk JC, Scheper GC. Vanishing white matter disease. Lancet Neurol 2006; 5:413. 4. Black DN, Booth F, Watters GV, et al. Leukoencephalopathy among native Indian infants in northern Quebec and Manitoba. Ann Neurol 1988; 24:490. 5. Upto date 6. Emedicine