Responsible Conduct of Research: Institutional Review Board
advertisement

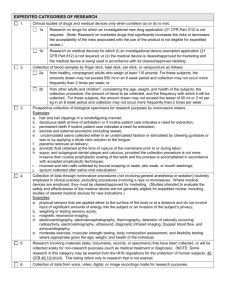
Responsible Conduct of Research: Institutional Review Board Office of Regulatory Research Compliance Objectives: To provide an overview of the Office of Regulatory Research Compliance and the role of institutional review committees; To describe role and function of the Institutional Review Board and the human subjects protection program; To discuss the IRB review process and the responsible conduct of research involving human participants. ABOUT THE OFFICE OF REGULATORY RESEARCH COMPLIANCE About The Office of Regulatory Research Compliance (ORRC) Location: The Office of Regulatory Research Compliance (ORRC) 1840 Seventh Street, NW HURB-1, Suite 309 Washington, DC 20001 202-865-8597(O) 202-232-5286 (Fax) Regulatory Committee Chairs Medical IRB • Thomas O. Obisesan, MD, MPH • Chief, Section of Geriatrics, HUH Non-Medical IRB • Broderick Eribo, PhD • Associate Professor, Biology Dept. IACUC • Thomas Heinbockel, PhD • Associate Professor, Anatomy Dept. IBC RMC • Stanley Tai, PhD • Associate Professor, Microbiology Dept. • Allan Johnson, PhD • Associate Dean, CNAHS Function of the ORRC ORRC supports HU in promoting ethical conduct of research and educating faculty and students about human subjects protections, responsible conduct of research, animal care regulations, and bio-safety. ORRC has oversight of all research approved by an institutional review committee to ensure regulatory compliance and the protection of all human and animal participants involved in research. Objectives: To provide an overview of the Office of Regulatory Research Compliance and the role of institutional review committees; To describe the role and function of the Institutional Review Board (IRB) and the human subjects protections program; To discuss the IRB review process and the responsible conduct of research involving human subjects. SPECIAL NOTE! NO HUMAN SUBJECTS RESEARCH, ANIMAL RESEARCH, OR RESEARCH INVOLVING BIOAGENTS AND RADIATION CAN BEGIN UNTIL IT HAS RECEIVED APPROVAL BY AN INSTITUTIONAL REVIEW COMMITTEE. ETHICAL FOUNDATION OF HUMAN RESEARCH PROTECTION Belmont Report Ethical Principles ETHICAL PRINCIPLES The “Belmont Report,” formally known as the Ethical Principles and Guidelines for the Protection of Human Subjects of Research, was written in 1979 by the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research. ETHICAL PRINCIPLES The Belmont Report identified the following three fundamental ethical principles that must be carefully considered to ensure the ethical practice of research involving human participants: Respect for Persons Justice Beneficence Respect for Persons • Participants have a right to informed consent. • Treat participants as autonomous agents capable of making an independent decision to enter into the research study voluntarily. • Additional provisions must be taken to protect participants that have a diminished capacity to act as an autonomous agent (children, prisoners, etc.). • Researcher should design procedures and safeguards which minimize the risk of invasion of privacy and assure confidentiality of data. Justice • No individual or group of participants should unduly bear the risks of research nor inequitably receive its benefits. • Fair distribution of risks and benefits of research. • Equitable selection of subjects.. • IRB has the responsibility of reviewing any requests by researchers to exclude selected subject populations. Beneficence • Do no harm. • Refers to the responsibility of the researcher to maximize possible benefits and minimize possible risks. • The benefits should outweigh the risks.. • IRB has separate applications for studies with “minimal risk” and “greater than minimal risk”. REGULATORY FOUNDATION OF HUMAN RESEARCH PROTECTION History Regulations Policies HIPAA History Significant recent events in research experimentation since the Hippocratic Oath. The Tuskegee Syphilis Study The Nuremberg Code The Milgram Study Declaration of Helsinki The Beecher Article The Syphilis Study Expose 1932 1947 1963 1964 1966 1970s The Code follows the Nazi experiments (1930s-40s) Yale psychologist Stanley Milgrim conducted a study to test this theory of obedience. “Ethics and Clinical Research Article” published citing 22 examples of human research being conduct not complying with Nuremberg Article appears in the New York Times in 1972 that caused the Tuskegee study to be shut down. History (cont.) The National Research Act The Belmont Report Consolidated HHS/FDA Regulations CIOMS Guidelines The Common Rule (45 CFR 46) National Bioethics Advisory Committee 1974 1978-79 1981 1982 1991 1995 HHS and FDA attempted to consolidate their regulations International Ethical Guidelines for Biomedical Research Involving Human Subjects prepared by the Council of International Organization for Medical Science Federal law that established the requirement of human subjects research review by IRBs * Recent findings on Guatemala and Henrietta Lacks. What regulations govern human subjects research? Department of Health and Human Services (DHHS) Food and Drug Administration (FDA) Sponsors Howard University Policies What regulations govern human subjects research? (cont.) All research involving human subjects must adhere to DHHS regulation 45 CFR 46, also known as the COMMON RULE, unless the requirement is waived by the IRB. DHHS 45 CFR 46 COMMON RULE 45 CFR 46: The Common Rule The Common Rule is a federal policy regarding Human Subjects Protection. The main elements include: (1) requirements for assuring compliance by research institutions; (2) requirements for researchers’ obtaining and documenting informed consent; (3) requirements for Institutional Review Board (IRB) membership, function, operations, review of research, and recordkeeping. Revisions under consideration affecting informed consent, security and data collection. http://www.hhs.gov/news/press/2011pres/07/20110722a.html What is HIPAA? The Health Insurance Portability and Accountability Act, commonly known as HIPAA, is another piece of legislation that impacts the conduct of human subjects research. Establishes national standards for the protection of private health information know as Protected Health Information (PHI). PHI is defined as any individually identifiable health information that is created or maintained by a Covered Entity (CE) department. HIPAA applies when: An investigator working in a Non CE department receives PHI from a CE department. An investigator working in a CE department creates, receives and/or discloses PHI for research purposes. The IRB is responsible for reviewing proposed HIPAA authorization forms, de-identification forms, and requests to waive the authorization process for research projects. ROLE OF THE IRB Basic IRB Member Responsibilities Conduct Protocol Review Applying Discipline Expertise and Regulatory Knowledge Attend Meetings Review Allegations of Noncompliance Maintain Confidentiality IRB REVIEW PROCESS Types of Protocol Submission Types of Protocol Review Criteria for Approval Criteria for Exemption; Expedited Review; Full Board Review Points to Consider (2 weeks) (4-6 weeks) (2-3 weeks) Types of Protocol Submission Examples of protocol submission: 1. 2. 3. 4. 5. 6. Initial Review Continuation Review Revisions/Modifications/Amendments Unanticipated Problems/Adverse Events Alleged or Reported Noncompliance Closures Mechanisms of IRB Review: Exempt Expedited Full • Research must meet the federal criteria for exempt review otherwise protocol will be forwarded for other either expedited or full board review • Outcome of the review may include any of the following: 1) approval; 2) request for additional information; 3) request for changes; 4) request that the review be conducted by the Full IRB. • Four possible outcomes: (1) approval; (2) minor revisions/additional information required (APPROVAL WITH ADMINISTRATIVE REVIEW); (3) major revisions/additional information required (TABLED); and (4) Disapproval What criteria must be met for research to be approved? Based upon the criteria for research approval set forth in the federal regulations 45 CFR Part 46.111, 50 CFR 56.111 and 38 CFR 16.111, the IRB should determine that all of the following conditions exist: • Risks to subjects are minimized • Risks to subjects are reasonable in relation to anticipated benefits, if any, to subjects • Selection of subjects is equitable. • Informed consent will be sought from each prospective subject • Informed consent will be appropriately documented • For greater than minimal risk, clinical research, or an NIH funded/FDA regulated clinical trial, the research plan makes adequate provision for monitoring the data collected to ensure the safety of subject. • There are adequate provisions to protect the privacy of subjects and to maintain the confidentiality of data. • Where any of the subjects are like to be vulnerable to coercion or undue influence, additional safeguards have been included in the study to protect subjects. Criteria for Exemption = 6: *Only the IRB can make the official determination that your project is exempt based upon 45 CRF 46.101 1 2 • Research conducted in educational settings involving normal educational practices on instructional strategies and teaching effectiveness • Research involving the use of educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures or observation of public behavior with certain conditions 3 •Research that is not exempt under paragraph b2 of this section, if: (i) the human subjects are elected or appointed public officials or candidates for public office; or (ii) federal statute(s) require(s) without exception that the confidentiality of the personally identifiable information will be maintained throughout the research and thereafter. 4 •Research involving the collection or study of existing data, documents, records, pathological specimens, or diagnostic specimens, if these sources are publicly available or if the information is recorded by the investigator in such a manner that subjects cannot be identified, directly or through identifiers linked to the subjects. 5 6 •Research and demonstration projects conducted by or subject to the approval of department or agency heads, and which are designed to study, evaluate, or otherwise examine certain conditions •Taste and food quality evaluation and consumer acceptance studies Criteria for Expedited Review Expedited reviewers must determine the following: Do the research activities meet the definition of “minimal risk” and do they fit within the federally mandated expedited categories? Do the research activities meet the eight federal criteria for IRB approval? “Minimal risk” is defined as “the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves from those ordinarily encountered in daily life or during the performance of routine physical or psychological examination or tests.” EXPEDITED REVIEW DOES NOT MEAN PUT A RUSH ON MY REVIEW BECAUSE A GRANT DEADLINE IS COMING UP!!! Criteria for Full Board Review • Research that does meet the criteria for exempt or expedited review must be submitted to the IRB for a review at a convened meeting at which a quorum of the members are present. • Medical IRB meets twice a month and the Nonmedical IRB meets at least once a month. • Research involving “greater than minimal risk” must go before the Full Review process. TYPICAL REVIEW TIMELINES REVIEW TYPE EXEMPT EXPEDITED RISK ASSESSMENT LOW OR LESS MINIMAL MINIMAL APPROVAL TIMELINE* 2 Weeks* 2-3 Weeks* FULL BOARD MORE THAN MINIMAL CONVENED FULL BOARD Review 4-6Weeks* *APPLICATIONS THAT REQUIRE REVISIONS MAY TAKE LONGER. Points to consider: How long? Exempt Review: 2 weeks Expedited Review: 2-3 weeks Full Board Review: 4-6 weeks Most proposals submitted require some form of revision. Submission of additional information will add time to your review and approval. Online CITI Training Module https://www.citiprogram.org IRB CHECKLIST Form B-1 (always required) Request for Expedited Review. Typed letter on letterhead signed by the PI. Request for Exemption (Form D-1). Note: One or both, depending on nature of the research Conflict of Interest Forms for all Investigators Certificate of completion of education in the protection of human research participants (required) Informed consent document(s) Signed copy of The Principal Investigator’s Assurance Form Any recruitment notices or advertisements Any survey instruments, psychological tests (other than standard, commercially available instruments), interview forms, or scripts to be used in the research Investigator’s qualifications (CV, biographical sketch) Formal research protocol, if available. Grant application, if applicable. SUBMIT ONE APPLICATION. TWO ADDITIONAL COPIES ARE NO LONGER REQUIRED! FORMS Applications forms are available online at: http://www.howard.edu/orrc Questions and Answers The Office of Regulatory Research Compliance Location: The Office of Regulatory Research Compliance (ORRC) 1840 Seventh Street, NW HURB-1, Suite 309 Washington, DC 20001 202-865-8597 (Office) Yonette F. Thomas, PhD Associate Vice President for Research Compliance