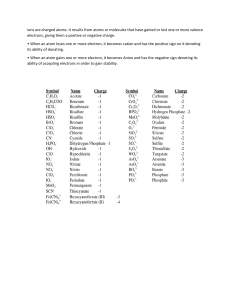
CHAPTER 2 SUBSTANCES are elements and compounds, mixtures are not. An ELEMENT is the simplest type of matter with unique physical and chemical properties. The LAW OF MASS COSERVATION states that the total mass of substances does not change during a chemical reaction. The LAW OF DEFINITE COMPOSITIONS states that no matter what its source, a particular compound is composed of the same elements in the same parts by mass. The mass fraction is the part of the compound’s mass that each element contributes. The sum of mass fractions equals 1. X = mass of element in compound/ mass of compound The LAW OF MULTIPLE PROPORTIONS states that if elements A and B react to form two compounds, the different masses of B that combine with a fixed amount of A can be expressed as a ratio of small whole numbers. In two compounds of the same elements, the mass fraction of an element relative to that of the other changes in increments based on ratios of small whole numbers. DALTON’S ATOMIC THEORY: • All matter consists of tiny indivisible atoms that cannot be created nor destroyed • Atoms of one element cannot be converted into atoms of another element • Atoms of an element are identical in mass and other properties and are different from atoms of any other element • Compounds result from the chemical combination of a specific ratio of atoms of different elements The cathode rays experiment proved that current was made of negatively charged articles which were then called electrons. Thomson measured the mass/charge ratio of these rays. Some years later, Millikan, with his oil-drop experiment, found the charge of this particle, which corresponds to -1.6 * 10 ^ -19 C. If atoms were neutral species containing negatively charged particles, they must also contain positively charged particles that counterbalanced the negative charge. Dalton proposed his plum pudding model based on these considerations. Rutherford, with his gold foil experiment, proved this model wrong and hypnotized a model where the positive nucleus was surrounded by a cloud of electrons spinning around it. An atomic nucleus consists of protons and neutrons which give the MASS NUMBER A. The number of protons alone is given by the ATOMIC NUMBER Z. Thus, N = A – Z. Isotopes are atoms of the same element which differ only in their number of neutrons. The atomic mass unit, now called Dalton, is 1/12 of the mass of C12. 1 Da = 1.66054 * 10 ^ -24 g [absolute mass] The isotopic composition of an element is determined by mass spectrometry. Relative abundance tells us which isotope is more common. The atomic mass is nothing but the average of the masses of its isotopes weighted according to their abundances. Atomic mass = ∑ (isotopic mass) * (fractional abundance of isotope) MONOATOMIC IONS: • N 3- nitride ion • Fe 2+ or 3+ • Cr 2+ or 3+ • Co 2+ or 3+ • Cu + or 2+ • Pb 2+ or 4+ • Hg2 2+ or Hg 2+ • Sn 2+ or 4+ POLYATMOIC IONS: • CH3COO- acetate • CN- cyanide • NO2- nitrite • NO3- nitrate • MnO4- permanganate • CO3 2- carbonate • HCO3- hydrogen carbonate/bicarbonate • CrO4 2- chromate • Cr2O7 2- dichromate • • • • • • • O2 2- peroxide PO4 3- phosphate HPO4 2- hydrogen phosphate H2PO4- dihydrogen phosphate SO32- sulfite SO42- sulfate HSO4- hydrogen sulfate/bisulfate ACIDS are considered as anions with one or more H+ added to them to give a neutral compound. BINARY ACID solutions form when gaseous compounds dissolve in water. Hydro- + nonmetal root + -ic + acid OXOACID names are similar to those of oxoanions except for the fact that -ate → -ic and -ite →-ous.IO2 iodite → HIO2 iodous acid | NO3 nitrite → HNO3 nitric acid | BrO4 perbromate → HBrO4 perbromic acid Ionic compounds don’t consist of molecules, so the mass of a formula unit is sometimes called formula mass instead of molecular mass. CHAPTER 3 The mole is the SI unit for the amount of substance. It is the amount of a substance that contains the same number of entities as the number of atoms in 12g of C12. This is the Avogadro number of particles (6.022 * 10^23) To determine the mass of an element from its mass fraction: Mass of element = mass of compound * (mass of element in 1 mol of compound / mass of 1 mol of compound). e.g. 15.5g NO2 → g O2 = 15.5g * [(2 mol * 16 g/mol) / (32 g/mol + 14 g/mol)] Isomers are compounds with the same molecular formula but different properties. They occur when atoms link together in different arrangements. To determine the empirical formula: 1. Determine the mass of each component 2. Determine the number of moles and write a preliminary formula 3. Convert the amount of moles to integers 4. To turn it into a molecular formula compare the molecular mass obtained with thee coefficients with the known molecular mass If instead you have mass percentages, assume 100.0 g of compound to express each mass percent directly as a mass. Reactions that occur in a sequence are often summarized into a single reaction. To d so, try to cancel the middle products. Example: 2Cu2S + 3O2 →2Cu2O + 2SO2 Cu2O + C → 2Cu + CO Cu2O has a coefficient of 2 in the first reaction so you multiply the second reaction by 2 and obtain: 2Cu2S + 3O2 + 2C → 2SO2 + 4Cu + 2CO The reaction yield is often less than 100%. It is calculated as actual yield / theoretical yield. If you have multistep syntheses just multiply the yields of each step together. CHAPTER 4 Water is a polar molecule in which the atoms are linked by polar covalent bonds. When an ionic compound dissolves in water, its atoms dissociate and are surrounded by the corresponding pole of water. This is called SOLVATION. When this happens, the solution’s electrical conductivity increases dramatically. A substance that conducts current when dissolved in water is an ELECTROLYTE. Concentration is an intensive property. It is most expressed as the MOLARITY: M = moles of solute / liters of solution [mol/L] Spectator ions are not involved in the actual chemical change, they are present only as part of the reactants. For example, in 2Ag + 2NO3 + 2Na + CrO4 → Ag2CrO4 + 2Na + 2NO3, Na and No3 are spectators; that is, we cannot add an Ag ion without also adding an anion (No3 in this case). Types of equations: MOLECULAR: 2KF + Sr(NO3)2 → 2KNO3 + SrF2(s) TOTAL IONIC: 2K+ + 2F- + Sr+ +2NO3- → 2K+ + NO3- + SrF2(s) NET IONIC: Sr+ +2F- → SrF2(s) In a PRECIPITATION REACTION, two soluble ionic compounds react to form an insoluble product: a precipitate. The electrostatic attractions between the ions outweigh the tendency of the ions to remain solvated and move throughout the solution. When the two solutions are mixed, the ions collide and stay together, and a solid product comes out. To know if a precipitate will form, remember that these are insoluble: • Chlorides, bromides and iodides of Ag, Pb, Cu and Hg2 • PbF2 and fluorides of group 2A • metal hydroxides • carbonates and phosphates • sulfides • CaSO4, SrSO4, BaSO4, Ag2SO4, PbSO4 Soluble ionic compounds are: • Compounds of group 1A • Ammonium compounds • Nitrates, acetates and perchlorates (NO3, CH3COO, ClO4) (SOLUBLE) • • • • • • Chlorides, bromides and iodides Fluorides Sulfates Group 1A hydroxides and Ca(OH)2, Sr(OH)2 and Ba(OH)2 Carbonates and phosphates of group 1A and NH4 Sulfides of group 1A, 2A and NH4 Reactions in which ions exchange partners are called double-displacement reactions or metathesis reactions. An acid-base reaction, also called a neutralization reaction, occurs when an acid reacts with a base. An ACID is substance that produces H+ ions when dissolved in water HX → H+(aq) + X-(aq) A BASE is a substance that produces OH- ions when dissolved in water MOH → M+(aq) + OH-(aq) Acidic solutions arise when a soluble ionic compound containing H+ ions dissociates in water. The dissociated H proton will form a covalent bond with one of the oxygen atoms of water, forming the hydronium ion H3O+. Acid and bases are characterized in terms of their strength. Strong bases and strong acids dissociate completely into ions, they are strong electrolytes; weak acids and bases do the opposite. STRONG ACIDS: HCl, HBr, HI, HNO3, H2SO4, HClO4 WEAK ACIDS: HF, H3PO4, CH3COOH STRONG BASES: group 1A hydroxides, heavy group 2A hydroxides (Ca, Sr, Ba); they have either OH- or O 2-as part of their structure WEAK BASES: NH3 do not contain OH- ions, but they all have an electron pair on a nitrogen atom. The key event in a strong acid – strong base reaction is that an H + ion from the acid and an OH- ion from the base form a water molecule. Only the spectator ions differ from one strong acid-base reaction to another. The ionic compound that results from the reaction of an acid and a base is called a salt. An acid and a base form a salt solution and water: HX + MOH → MX + H2O The cation of the salt comes from the base and the anion comes from the acid. Acid-base reactions are metathesis. STRONG ACID + STRONG BASE → the strong acid in water is actually solvated H3O+ and anions (HX + H2O → H30+ + X-). If we add a strong base, the hydronium ion transfers a proton to OH, leaving a water molecule. (H3O+ + X- + M+ + OH- → MX + H2O). An acid-base reaction is a proton-transfer process, H3O+ acts as an acid and donates a proton to OH-, which act as the base. ACID + CARBONATE → when an ionic carbonate reacts with an acid, one of the products is carbon dioxide. 2HCl + K2CO3 → KCl + [H2CO3] (highly unstable) [H2CO3] → H2O + CO2 Writing the total ionic equation… 2HCl + 2H2O → 2H3O+ + 2Cl2H3O++ 2Cl-+ 2K++ CO32-→ 2Cl-+2K++ 3H2O(l) + CO2(g) + 2There is a proton transfer from H3O to CO3 . Ionic sulfites react similarly, with the formation of gaseous SO2. WEAK ACID + STRONG BASE → the acid is weak and dissociates very little, so it appears on the left side of the equation as an intact molecule, instead of as ions. The proton transfer happens directly from the weak acid rather than from the hydronium ion. There is only one spectator, which is the cation from the strong base. The net ionic equation is HX(aq) + (M)OH-(aq) → (M)X- + H2O(l) Weak acids also react with carbonates to form gaseous Co2, but the weak acid is written as an intact molecule instead of as ions. Acid-base reactions are studied in a procedure called titration, which consists in calculating the unknown concentration of acid through the known concentration of a standardized base. It is made of two key stages: 1. The equivalence point occurs when the amount of H + ions in the original volume of acid reacts with the same amount of OH- ions from the buret 2. The end point occurs when a tiny excess of OH- ions changes the indicator (the substance mixed with the acid, whose color is different in acid than in base) permanently to its basic color. In calculations we assume that this tiny excess of OH- ions is insignificant and that the amount of base needed to reach the two key stages is the same. OXIDATION is the loss of electrons, the element which loses electrons is the reducing agent; REDUCTION is the gain of electrons, the element which gains electrons is the oxidizing agent. The oxidizing agent is reduced, the reducing agent is oxidized. CHAPTER 5 BOYLE’S LAW CHARLE’S LAW ! The Kelvin scale must be used. Combining the two we get 𝑃𝑉 𝑇 =𝑘 AMONTON’S LAW is a consequence of the two: P = kT at constant volume and n. AVOGADRO’S LAW Equal volumes of different gases contain the same number of particles, at constant P and T. Combining these laws we get the ideal gas law PV = nRT R = 0.0821 (atm * L)/(mol*K) = 8.314 (Pa*m^3)/(mol*K) d = m/V = (n*M*P)/(n*R*T) = MP/RT M = (mRT)/(PV) Gases mix homogeneously in any proportion and each gas in a mixture behaves as if it were the only gas present. DALTON’S LAW OF PARTIAL PRESSURES In a mixture of unreacting gases, the total pressure is the sum of the partial pressures of the individual gases. Ptot = ∑Pi so Ptot * Vtot = ntot * RT Each component contributes a farction of the total number of moles in the mixture, the mole fraction X. The sum of the mole fractions must be 1. X = ngas/ ntot Pgas = Xgas * Ptot Whenever a gas is in contact with water, some of the water vaporizes into the gas. The water vapour that mixes with the gas contributes a pressure known as vapour pressure and depends only on water temperature. The KINETIC-MOLECULAR THEORY (Maxwell, Boltzmann) accounts for macroscopic gas behaviour at the level of individual particles. It is based on three postulates: 1. Each particle is a point of mass (volume too small compared to the container) 2. Particles are in constant, straight-line, random motion except when they collide 3. Collisions are elastic, there is no loss of Ek through friction (the kinetic energy is constant). The curves flatten as the temperature increases, which means that the uncertainty in the particles speed increases. The peak of each curve increases as the temperature increases, which means that increasing T the most probable speed increases too. This is because the average kinetic energy is proportional to T, through the relationship Ek avg = k * T This means that, at a given T, all gases will have the same average Ek. Lighter molecules move faster, on average, than heavier molecules. Ek = (mv^2)/2 -> a slower and heavier object can have the same Ek of a faster and lighter one. Because the molecules of any gas hit the walls with the same Ek, at a given T, equimolar samples of any gases exert the same pressure and, thus, occupy the same volume. If we equal this formula and the classical formula for Ek, and solve for the speed, we get the root-mean-square speed: a particle moving at this speed has the average kinetic energy. We use R equal to 8.314 and M in kg/mol. EFFUSION is the process by which a gas escapes through a tiny hole in its container into an evacuated space. Graham’s law of effusion: the rate of effusion of a gas is inversely proportional to the square root of its molar mass. Since the smaller the molar mass the higher the urms, more atoms of the lighter gas will escape per unit time. The different rates are used to separate uranium isotopes to produce nuclear weapons. DIFFUSION is the movement of one gas through another, the formula does not change. Diffusion rates are much lower. The mean free path is the average distance a particle travels without collisions. Divide the most probable speed by the mean free path and you’ll get the collision frequency. As pressure and temperature conditions become more extreme, the interparticle attractions become more important. At normal Pext, the spaces between the particles are so large that their volume can be neglected. As Pext rises, V decreases and the particles get closer, their volume cannot be ignored so the numerator of PV/RT is smaller. Also, interparticle attractions have a greater effect: they lessen the impact force and cause a pressure decrease, so we’ll have a lower value of PV/RT. Note that He and H2 don’t show any deviation because of their weak interparticle attractions. Van der Waals equation: a depends on the number and distribution of electrons, b depends on particle volume. CHAPTER 6 Internal energy: U = Ep + Ek ; ∆U = q + w or ∆U = ∆H - ∆nRT , where ∆n is the difference between the sum of the coefficients of the reactants and the products. A change in the energy of the system must be accompanied by an equal and opposite change in the energy of the surroundings. The first law of thermodynamics states that the total energy of the system is constant, that is, energy cannot be created nor destroyed: ∆U universe = ∆U system + ∆U surroundings = 0. Internal energy is a state function: it depends only on the final and initial states. The enthalpy is H = U + PV → ∆H = ∆U + P∆V → ∆H = q + w + P∆V , but q = -∆PV, so ∆H = q AT CONSTANT P It is a state function too: ∆H = H products – H reactants. ∆H < 0 exothermic process, ∆H > 0 endothermic process. The heat capacity is the quantity of heat required to change the temperature of a substance by 1 degree. C = q / ∆T [J/K] [1 cal = 4.184 J 1kJ = 239 cal] The specific heat capacity is the quantity of heat required to change the temperature of 1 gram of substance by 1 degree: c = q / (∆T * mass) → q = c * mass * ∆T The molar heat capacity is is the quantity of heat required to change the temperature of 1 mol of substance by 1 degree: c = q / (∆T * n) [J/mol*K] The magnitude of ∆H is proportional to the number of moles. We use ∆H to convert an amount (mol) of a substance to an equivalent quantity of heat and vice versa. HESS’S LAW states that the overall enthalpy change is the sum of the enthalpy changes of individual steps. ∆H overall = ∆H1 + ∆H2 + … + ∆Hn Calculating an unknown ∆H involves three steps: 1. Identify the target equation, the step whose ∆H is unknown and note the amount of each reactant and product 2. Adjust each equation with known ∆H so that the target amount is on the correct side of the equation (change the sign of ∆H when you reverse the equation; multiply mol and ∆H by the same factor) 3. Add up the adjusted equations and their resulting ∆H. All substances except those in the target equation must cancel. Chemists have established a set of specific conditions called standard states: • For a gas, the standard state is 1 bar and ideal behaviour • For a substance in aqueous solution, standard state is 1M concentration • For a pure substance the standard state is the most stable form of the substance at 1 atm and the temperature of interest (usually 25 °C) Standard states are indicated by °. The standard enthalpy of reaction is ∆H°rxn. For an element in its standard state, ∆H°formation = 0. For most metals, the standard state is the solid one. For mercury and bromine the standard state is liquid. For molecular elements, the standard state is the molecular form (Cl2, H2…). Some elements exists in different forms, called allotropes, but only one is the standard state ( C graphite, O2, S8). Most compounds have negative ∆H°f. The standard enthalpy of reaction is the sum of the standard enthalpies of formation of the products minus the sum of the standard enthalpies of formation of the reactants. ∆H°rxn = ∑m∆H°f products - ∑n∆H°f reactants, where m and n are the mol of the products and reactants given by the coefficients in the balanced equation. CHAPTER 7 v= υ*λ Refraction happens when a wave strikes an object at an angle other than 90° and changes speed and direction. Diffraction happens when a wave passes through a slit and bends around both edges, forming a semicircular wave on the other side of the opening. If the slits are two and adjacent, there can be a constructive or a destructive interference. BLACKBODY RADIATION, light given off by a hot black body (an idealized object that absorbs all the radiation). To find an explanation, Planck assumed that the object could emit or absorb only certain quantities of energy E = nhυ, where n is a quantum number and h is the Planck’s constant: h = 6.626 * 10^-34 J*s (kg m^2 / s) Since energy could only be emitted by the object’s atom, atoms emit only certain quantities of energy, thus the energy of an atom is quantized. Each energy packet is called a quantum and it is equal to hυ. ∆E = ∆nhυ. The smallest change occurs when an atom changes to an adjacent energy state (∆n = 1) Combining the equations: ∆E = hυ = hc / λ THE PHOTOELECTRIC EFFECT when monochromatic light of sufficient frequency shines on a metal, a current flows. For current to flow, the light must have a threshold frequency, different for every metal. It was thought that an electron would break free when it absorbed enough energy from light of any color, provided that the amplitude of its wave was big enough. In reality, the energy of a wave is associated to its frequency, not to its intensity. Einstein proposed that light itself is particulate and quantized into bundles of energy called photons. Each atom changes its energy by an amount ∆E when it absorbs or emits one photon. The intensity is related to the number of photons, not to its energy, which is why also dim light causes the effect without any time lag. When light from electrically excited gaseous atoms passes through a slit and is refracted by a prism, it does not create a continuous spectrum, but a line spectrum: a series of lines separated by black spaces. Rydberg developed the Rydberg equation, which predicts the position and wavelength of any line in a given series: R = 1.0967 * 10^-7 m^-1, n2 > n1 To calculate the lines in the visible series, n1 = 2. A major problem arose: the electron would have to spin around the nucleus not to collapse on it, but in order to do so it would have to emit radiation and lose energy, spiraling into the nucleus. Bohr proposed a model for the H atom that did predict the existence of line spectra: 1. The H atom has only certain stationary states, associated with a fixed circular orbit around the nucleus 2. The atom does not radiate energy while in one of its stationary states 3. The atom changes to another stationary state only by absorbing or emitting a photon, whose energy equals the difference in the energies of the two states. The quantum number n is associated to a specific orbit radius and the smaller the radius, the lower the energy level. When the electron is in the first orbit, it is in the ground state. Otherwise it is in an excited state. Since there are only certain energy levels in the atom, only certain quantities of energy can be emitted → discontinuous atomic spectrum When electrons in the H atom drop to n=3, photons with the energy of infrared radiation are emitted; if n = 2 visible radiation, if n = 1 ultraviolet. To calculate the energy levels of an atom: E = -2.18 * 10^-18 J (Z^2/n^2), where Z is the charge of the nucleus We can use it to calculate the difference in energy between two levels. When the atom emits energy, the electron moves closer to the nucleus, so n final < n initial and E is a larger negative number and ∆E < 0. When the atom absorbs energy, the electron moves away from the nucleus, so n final > n initial so ∆E> 0 and E is a smaller negative number. We can find the ionization energy, the energy required to form 1 mol of gaseous H+ ions from 1 mol of gaseous H atoms, putting n final = infinity and multiplying by Na. Once we know ∆E we can find the wavelengths of the spectral lines from λ = hc / ∆E, from which we can derive Rydberg’s equation. De Broglie proposed that if energy is particle-like, then perhaps matter is wavelike: if electrons have wavelike motion in orbits of fixed radii, only certain frequencies and energies are allowed. Combining E = mc2 (particle) and E = hv = hc / λ (wave) → λ = h / mu (De Broglie wavelength) Heavy objects have wavelengths many orders smaller than the objects themselves, too small to be detected. If electrons have properties of energy, do photons have properties of matter? We can calculate a photon’s momentum ( mass * speed) using De Broglie equation: mu = P = h / λ. A decrease in the momentum should lead to an increase in wavelength. Compton directed a beam of x-rays at graphite and observed an increase in the reflected photons wavelength. Thus, photons transfer momentum to electrons when they collide. Heisenberg postulated the uncertainty principle: ∆x * m * ∆u >= h / 4π For a macroscopic object, the uncertainties are insignificant because the mass is enormous compared with h / 4π. The Schrodinger equation: E is the energy, psi is the wave function, or atomic orbital. The Hamiltonian operator is a set of mathematical operations that yields one of the allowed energy states of the atom. Each solution 2 gives an energy state associated with a given atomic orbital. Ψ is the probability density and it is a measure of the probability of finding the electron in some tiny volume of the atom. The difference in energy between two consecutive levels is: An atomic orbital is specified by three quantum numbers: 1. Principal quantum number n: size of the orbital and energy level 2. Angular momentum quantum number l: from 0 up to n-1, shape, indicates the sublevel. l=0 →s l =1→p l= 2→d l = 3→f 3. Magnetic momentum quantum number ml: from -l up to l, orientation in space (2l+1 = number of orbitals for a given l). The total number of ml values for a given n is n2= total number of orbitals in energy level. CHAPTER 8 Exclusion principle: no two electrons in the same atom can have the same four quantum numbers → an atomic orbital can hold a maximum of two electrons, which must have opposite spins. Higher charges interact more strongly than lower charges, therefore, a higher Z increases nucleus-electron attractions and, thus, lower sublevel energy (atoms more stable). Shielding by other electrons which repel each other somehow reduces the effect of the full nuclear charge to an effective nuclear charge Zeff, the nuclear charge an electron actually experiences, and this lower nuclear charge makes the electron easier to remove. Electrons in the same energy level shield each other. Inner electrons shield outer ones very effectively. Penetration increases nuclear attraction for a 2s electron over that for a 2p electron and decreases the shielding of a 2s electron by the 1s electron. Since it takes more energy to remove a 2s e- than a 2p, the 2s sublevel is lower in energy than the 2p. Penetration splits an energy level into sublevels of differing energy: the lower l, the more its electrons penetrate and so the greater their attraction to the nucleus → lower l = more stable (lower energy) sublevel. Aufbau principle: in the ground state of an atom or ion, electrons fill atomic orbitals of the lowest available energy levels before occupying higher levels. Hund’s rule: when orbitals of equal energy are available, the electron configuration of lowest energy has the maximum number of unpaired electrons with parallel spins. In any period, the ns sublevel fills before the (n-1)d sublevel. However, chromium has one electron in the 4s sublevel and 5 in the 3d, making both half-filled, and copper has one electron in the 4s sublevel and 10 in the 3d. Metallic radius: ½ the shortest distance between the nuclei of adjacent atoms in a crystal of the element: Covalent radius: ½ the shortest distance between nuclei of boded atoms. ATOMIC SIZE As n increases, atomic size increases. As Zeff increases, the atomic size decreases. Down a group n dominates, across a period electrons are added to the same outer level, so the shielding does not change. Zeff rises ad atomic radius decreases. IONIZATION ENERGY It is the energy required for the complete removal of 1mol of electrons from 1mol of gaseous atoms or ions. It is always positive. Atoms with a low IE tend to form cations. As size decreases it takes more energy to remove an electron, so IE decreases down a group (except from aluminum down) and increases across a period (except for Al and O and S). ELECTRON AFFINITY It is the energy change (release) accompanying the addition of 1mol of electrons to 1 mol of gaseous atoms or ions. The second EA is always positive because energy must be absorbed to overcome electrostatic repulsions and add another electron to a negative ion. METALLIC BEHAVIOUR Metals ate typically shiny solids; they are good conductors and lose electrons to nonmetals. ACID-BASE BEHAVIOUR (both = amphoteric) Metals → ionic oxides → bases in water Nonmetals →covalent oxides → acids in water. Species with all of their electrons paired exhibit diamagnetism: they are not attracted by a magnetic field. Otherwise, they are paramagnetic. CHAPTER 9 IONIC BONDING Energy is first absorbed during electron transfer and then released to form a lattice. The lattice energy is the enthalpy change that accompanies the separation of 1mol of ionic solid into gaseous ions. It is not measured directly, it is determined using Hess’s law in a Born-Haber cycle, a series of steps from elements to ionic solid for which all the enthalpies are known, except the lattice energy. The steps are hypothetical, not the actual ones. Ionic solids exist only because the lattice energy far exceeds the total energy needed to form the ions. Lattice energy is directly proportional to electrostatic energy (Coulomb): Ionic size ↑ lattice energy ↓ Ionic charge ↑ lattice energy↑ The electron-sea model of metallic bonding proposes that all the metal atoms in the sample contribute their valence electrons to form a delocalized electron sea throughout the piece, with the metal ions lying in an orderly array. All the atoms in the sample share the electrons and the piece is held together by the mutual attraction of the metal cations for the mobile valence electrons. COVALENT BONDING A covalent bond arises from the balance between the nuclei attracting the electrons and electrons and nuclei repelling each other. The bonding order is the number of electron pairs being shared by a pair of atoms. The bond energy is an average of the energy needed to overcome the attraction between the nuclei and the shared electrons. It is the standard enthalpy change for breaking the bond in 1 mol of gaseous molecules. It is an endothermic process, so BE > 0. A higher bond order results in a smaller bond length and as the bond length decreases the BE increases. Covalent substances melt and boil at low temperatures because of weak intermolecular forces, not intramolecular. Some molecules have covalent bonds in three dimensions, throughout all the sample, and these molecules do reflect the strength of covalent bonds (quartz, diamond). The heat released or absorbed during a chemical change is due to differences between reactant bond energies and product bond energies. Bond breaks → energy absorbed. Bond forms → energy released. ∆H°reaction = ∑∆H°bonds formed + ∑∆H°bonds broken Electronegativity is the ability of a bonded atom to attract shared electrons. It is inversely related to atomic size. According to Mulliken definition, EN = (EA + IE) / 2. O.N. = valence e- - (unshared e- + shared e-). CHAPTER 10 Place the atom with the lowest group number in the middle. Usually this is also the atom with the lowest electronegativity. If the atoms have the same group number, place the atom with the higher period number in the centre. H is never a central atom. If a central atom does not end up with an octet, form more bonds. Resonance structures have different locations of bonding and lone pairs; this is due to electron-pair delocalization. This leads to fractional bond orders: shared electron pairs (bonds) / number of atoms among which the bonds are shared. Calculate the formal charge: the charge it would have if the electrons were shared equally: f.c = shared e- - (unshared e- + ½ shared valence e-) = valence e- - lone e- - ½ bonding e- Only when an atom has an f.c. = 0 it has its usual number of bonds. The best resonance structure is the one with 1) smaller f.c. 2) the same f.c. (except 0) are avoided on adjacent atoms 3) the most negative f.c. should be on the most electronegative atom. EXCEPTIONS TO THE OCTET RULE In gaseous molecules containing Be or B as the central atom (BCl2 or BF3), it is often electron deficient: it has fewer than 8 electrons around it. They don’t form multiple bonds because the halogens are much more electronegative. Free radicals have an odd number of valence electrons and contain a lone e- which makes them paramagnetic. They are dangerous because they can bond to an H atom in a biomolecule and extract it, forming a new free radical. Antioxidants like vitamin E interrupt free-radical proliferation. !! The most important resonance structure of NO2 is that on the left: Many molecules have more than eight valence electrons around the central atom. Expanded valence shells only occur from period 3 on because they have d orbitals available. The expansion releases energy (more bonds are formed). Examples are PCl5 and SF6, SF4, ClF3 (even though the octet is not reached with bonds). VSEPR THEORY Valence-shell electron-pair repulsion (VSEPR) theory: to minimize repulsions, each group of valence electrons around a central atom is located as far as possible from the others. A group can be: a single, double, triple bond; a lone pair; a lone electron. Only electrons around the central atom affect shape. Both shape and polarity determine molecular polarity, an uneven distribution of charge. Dipole moment μ measure this polarity in Debye (D): 1D = 3.34 * 10^-30 C*m. CHAPTER 11 VB THEORY: a covalent bond forms when orbitals of two atoms overlap and sp a pair of electrons occupy the overlap region. Their wave functions are in phase. The space formed by the overlapping orbitals has a maximum capacity of two paired e-. The greater the overlap, the closer the nuclei are to the eand the stronger the bond. A p or d orbital is oriented so as to maximize overlap. Pauling proposed that the valence orbitals in the isolated atoms become new orbitals. The number of hybrid orbitals equals the number of atomic orbitals mixed. The shape and orientation of such orbitals maximizes the overlap region. sp hybridization: two sp2 sp3 nonequivalent orbitals of a central atom, one s and one p, mix and form two equivalent sp hybrid orbitals that are oriented 180° apart (linear shape). sp2 hybridization: one s + two p → three sp2 (trigonal planar and bent shapes). sp3 hybridization: one s + three p → four sp3 (tetrahedral shape). sp3d hybridization: one 3s, three 3p and one of the five 3d orbitals → five sp3d (trigonal bipyramidal shape) sp3d2 hybridization: one 3s, three 3p, two 3d → six sp3d2 (octahedral shape) Hybridization does not apply to large nonmetal hydrides (H2S) because larger atoms form such long bonds with H that repulsions are small and overlap of UNHYBRIDIZED atomic orbitals happens. It does not apply to expanded valence shells either because d orbitals have such high energies that a combination of hybridized sp orbitals and unhybridized 3p orbitals is better. A sigma bond is formed by end-to-end overlap of orbitals. It has its highest electron density along the bond axis. All single bonds are σ bonds. A pi bond is formed by side to side overlap of orbitals. It has two regions (lobes) of electron density, one above and one below the σ-bond axis. The two electrons in one π bond occupy both lobes. A double bond consists of one σ bond and one π bond, which increases electron density between the nuclei. A pi bond is weaker than a sigma bond. Sigma bonds allow rotation, pi bonds don’t. For this reason cis and trans structures exist for some compounds. MO THEORY: just as an atom has atomic orbitals, a molecule has molecular orbitals. The theory pictures a molecule as a collection of nuclei with orbitals that extend over the whole molecule and are occupied by delocalized electrons. It requires some approximations: to mathematically combine (add or subtract) atomic wave functions of nearby atoms to form molecular wave functions. Adding them forms a bonding MO, which has a region of high electron density between the nuclei; subtracting them forms an antibonding MO, which has a node where the electron density is zero between the nuclei. The number of AOs combined always equals the number of MOs formed. A bonding MO is lower in energy than the AO that forms it. Because the electron density is spread mostly between the nuclei, nuclear repulsions decrease while nucleus-electron attractions increase. Moreover, electrons can delocalize their charges over a larger volume, lowering electron repulsions. An antibonding MO is higher in energy than the AOs that form it. Nuclear repulsions increase and the molecule is less stable. For AOs to interact and form Mos, they must be similar in energy and orientation. BO = (e in bonding MOs - e in antibonding MOs) / 2 BO > 0: the molecule is more stable than the separate atoms, so it will form. BO = 0: the molecule is as stable as the separate atoms, so it will not form. The higher the BO, the stronger the bond. Homonuclear diatomic molecules (H2, O2, N2) are composed of two identical atoms. MOs from p-orbitals combination can be: • Sigma MOs, bonding and antibonding • Pi MOs, bonding and antibonding Atomic orbitals overlap more extensively end to end than side to side. Thus, the σ2p MO is usually lower in energy than the π2p MO. σ2p < π2p < π*2p < σ*2p B, C, and N atoms are relatively large, with 2p AOs only half-filled, so repulsions are weaker. As a result, orbital energies are close enough for some mixing to occur between the 2s of one atom and the end-on 2p of the other. The effect is to lower the energy of the σ2s and σ*2s MOs and raise the energy of the σ2p and σ*2p MOs; the π MOs are not affected. π2p < σ2p < π*2p < σ*2p Orbital occupancy correlates with paramagnetism. Oxygen has two unpaired electrons in the π*2p antibonding MO and is paramagnetic. Heteronuclear diatomic molecules have asymmetric MO diagrams. Atoms with greater Zeff pull their electrons closer, so they have lower energy AOs and higher EN values. HF: the high Zeff of F means that the 1s, 2s, and 2p orbitals have lower energy than the 1s of H. The half-filled 2p orbital of F interacts with the 1s of H. The two filled 2p orbitals of F are called nonbonding MOs and they have the same energy as the isolated AOs. In polar covalent molecules, bonding MOs are closer in energy to the AOs of the more electronegative atom. CHAPTER 12 The potential energy in the form of intermolecular forces tends to draw molecules together; the kinetic energy associated to motion tends to draw them apart. The physical state of a substance depends on the interplay between the two: in a gas, the potential energy is small relative to the kinetic energy; thus, on average, the particles are far apart. In a liquid, attractions are stronger because the particles are touching, but they have enough kinetic energy to move around each other. In a solid, the attractions dominate the motion. Condensing, freezing and deposition are exothermic changes; melting, vaporizing and sublimation are endothermic. ΔH°fus < ΔH°vap because it takes more energy to completely separate the molecules from each other. ΔH°subl = ΔH°fus + ΔH°vap ΔH°cond = - ΔH°vap and so on… Heat not during a phase change: Q = n * C [J/mol*K] * ΔT [remember that C = mass * specific heat] Heat during a phase change: Q = n * ΔH°phase change The kinetic energy stays the same (T is constant), it is the potential energy which gets reduced / increased. The total heat change is the sum of the heat changes of all the stages. Dynamic equilibrium: molecules enter and leave the liquid at equal rates. With time, the number of molecules colliding with and entering the surface—the rate of condensation—increases as the vapor becomes more populated, so the increase in pressure slows. Eventually, the rate of condensation equals the rate of vaporization and the pressure is constant. The pressure exerted by the vapour at equilibrium is called vapour pressure. The higher the T, the higher the vapour pressure. The weaker the intermolecular forces, the higher the vapour pressure. The nonlinear relationship between vapour pressure and temperature is converted into a linear one with the Clausius-Clapeyron equation: (R = 8.314 J/mol*K), C is a constant, NOT heat capacity. Boiling point: the temperature at which the vapor pressure inside bubbles in the liquid equals the external pressure, which is usually that of the atmosphere. Higher P → higher boiling point (the particles need more kinetic energy to form bubbles) Melting point: the temperature at which the melting and freezing rate are equal. As more molecules enter the liquid (molten) phase, some collide with the solid and become fixed in position again. In general, the solid is stable at low T and high P, the gas at high T and low P. The solid-liquid line has a positive slope because the solid is denser than the liquid. For water, instead, it is the opposite, water expands upon freezing, and the slope is negative. The three phases meet at the triple point, where they are at equilibrium. CO2 does not melt at ordinary conditions because in order to do so we must have a pressure of 5.2 atm. At the critical point the density of the vapour and that of the liquid are equal, the phase boundary disappears. The average Ek is so high at this point that the vapour cannot be condensed at any pressure. Beyond the critical temperature a supercritical fluid (SCF) is formed. It has unusual solvent properties. INTERMOLECULAR FORCES The distance between two nonbonded atoms in adjacent molecules is called the van der Waals (VDW) distance, at which intermolecular attractions balance electron-cloud repulsions. The van der Waals radius is one-half the closest distance between nuclei of identical nonbonded atoms. It is always larger than its covalent radius. Like covalent radii, VDW radii decrease across a period and increase down a group. • Ion - dipole forces. Like when an ionic compound dissolves in water. • Dipole - dipole forces. The positive pole of a molecule attracts the negative pole of the other one. They depend on the magnitude of the dipole moment. • Ion-induced dipole and dipole-induced dipole occur when an ion or a dipole induce a dipole in a polarizable electron cloud. How easily this can happen is called polarizability. It increases down a group and decreases across a period. Cations are less polarizable than their parent atoms, anions the opposite. • Hydrogen bond. When an H atom is bonded to a highly electronegative one with lone pairs (O, N or F). The H-N, H-O and H-F bonds are very polar and when the partially positive H of one molecule is attracted to the partially negative lone pair on the N, O or F of another molecule, a hydrogen bond forms. It takes more energy for the molecules to separate and enter the gas phase, so the boiling point increases. • London forces. They result from the motion of electrons in atoms. This motion causes a variation of e concentration which induces a dipole in an atom that is close. They are instantaneous dipole-induced dipole forces. This process spreads throughout the sample. At low T, these attractions keep the atoms together. They are the only forces existing between nonpolar molecules and exist between all particles. They are the dominant forces even in polar molecules. Polarizability depends on the number of electrons, so larger molecules will have stronger London forces and higher boiling points. For a pair of nonpolar substances with the same molar mass, a molecular shape that has more area of contact allows stronger attractions. THE LIQUID STATE An interior molecule is attracted by others on all sides; a surface molecule is only drawn downwards and therefore tends to move towards the interior. For this reason, a liquid surface has the fewest molecules and, thus, the smallest area possible. The only way to increase the area is for molecules to move up by breaking attractions in the interior. Surface tension: the energy required to increase the surface area by a given amount and is measured in J/m^2. The stronger the intermolecular forces, the greater the surface tension. It decreases with increasing temperature. Capillarity: the rise of a liquid against the pull of gravity through a narrow space. It is the result of a competition between intermolecular forces within the liquid and those between the liquid and the tube walls. Viscosity: the resistance of a fluid to flow, due to intermolecular forces which impede the movement of molecules around and past each other. It decreases with higher T and it is affected by molecular shape. WATER It dissolves ionic compounds through ion-dipole forces, polar nonionic substances like ethanol CH3CH2OH and glucose by H bonding, nonpolar gases to a limited extent through dipole-induces dipole and dispersion forces. It has a high specific heat capacity, a high heat of vaporization, high surface tension and high capillarity, all due to H bonding. The large spaces within ice make the solid less dense than the liquid. As pressure is applied, some H bonds break so the crystal structure is disrupted and the ice liquefies. It is most dense at 4°C. SOLIDS Crystalline solids: have well-defined shapes because their particles are orderly arranged Amorphous solids: have poorly defined shapes because their particles are not orderly arranged Crystals are arranged in a lattice: all points with identical surroundings. The smallest portion is called unit cell. There are three types of unit cell: 1) Primitive unit cell: the centres of eight identical particles define the corners of a cube and the particles touch at the edges. Each contains 1 atom (1/8 * 8). Coordination number is 6. 2) Body-centred cubic unit cell: same + one atom in the centre. Total 2 atoms, coordination number is 8 3) Face-centred cubic unit cell: at the edges (1/8 * 8) + one in the centre of each face (1/2 * 6). None at the centre of the cube. Total 4 atoms. C.N. = 12. There are 14 Bravais lattice, the most common are cubic (alpha = beta = theta = 90°) and hexagonal (alpha = beta = 90° != theta = 120°). Noble gases are the only ones that form atomic solids: solids held together only by dispersion forces. Molecular solids are solids in which individual molecules occupy the lattice points. To maximize attractions in a binary ionic solids, cations are surrounded by as many anions as possible, and vice versa. The unit cell has the same cation/anion ratio as the empirical formula. Lattice energy is here very strong: they have high melting points and low electrical conductivities. If enough force is applied, the crystal cracks. In Na+ Cl-, each Na+ is surrounded by 6 Cl-, the coordination number is 6: 4 Cl- + 4 Na+ = 1/1 ratio cations/anions. Metallic solids are held together by strong metallic bonds. Their properties result from delocalized electrons. Network covalent solids are linked together by strong covalent bonds: - Graphite is the stable form of C and occurs as stacked flat sheets of hexagonal carbon rings with a strong sigma bond framework and delocalized pi bonds. The electrons are delocalized over the entire sheet: graphite is a good conductor but only in the plane of sheets, which interact via dispersion forces and impurities between the sheets allow them to slide past each other. - Diamond: adopts a face-centred cubic unit cell. It does not conduct electricity because the bonding electrons are delocalized. Polymorphism: a compound may occur in different crystalline forms, like SiO2 (quartz) Allotropy: it is the same but for elements, like carbon. Other C forms are: nanotubes (strong mechanical properties, very light) and fullerenes. Band theory: the energies of the Mos formed are so close that they form a continuum, or band, of MOs. The lower energy MOs make up the valence band. The empty MOs are the higher in energy and make up the conduction band. These two bands are contiguous: they highest level of one touches the lowest one of the other. This means that, given an infinitesimal quantity of energy electrons jump from the filled valence band to the unfilled conduction band: they are delocalized and free to move. That’s why metals conduct current so well. Lustre is due to the fact that electrons absorb and release photons of many frequencies as they move between the bands. In a conductor, conductivity decreases with T; in a semiconductor it increases. Superconductivity is the conduction without any energy loss (electrons collide with vibrating atoms and lose energy). If we assume T = 0K (no atom vibrations) we can calculate the Fermi level, the highest occupied level. 1 electron Volt is ∆E = 1eV ∆E = 5eV the kinetic energy gained by an electron accelerated from rest through a potential of ∆V = 1V. E = q * ∆V = 1.602* 10^-19 C * V (Joule). Conductivity in a semiconductor can be greatly enhanced by doping: • n-type semiconductor: when Si is doped with P, which contains one valence e- more, the additional eenters an empty orbital in the conduction band, bridging the energy gap and increasing conductivity. Extra negative charges are present; • p-type semiconductor: when Si is doped with Ga, which contains one valence e- less, since some of the orbitals in the valence band are empty, Si electrons can migrate to these empty orbitals, increasing conductivity. In contact wit each other, they form a p-n junction. CHAPTER 13 The solubility is the maximum amount that dissolves in a fixed quantity of a given solvent at a given temperature when an excess of solute is present. Likes dissolves like because the forces within solute and solvent can replace each other. Thus: salts are soluble in water, insoluble in hexane (C6H14); oil is insoluble in water, soluble in hexane. Alcohols have a dual polarity: a polar OH group bonded to a nonpolar hydrocarbon group. The OH part interacts through strong H bonds with water and the hydrocarbon portion interacts with hexane through dispersion forces. For gases, a higher boiling point indicates stronger intermolecular forces and should result in greater solubility in water. When a gas dissolves in a solid, it occupies the spaces between the closely packed particles. Solid-solid solutions are usually heterogeneous. Alloys are solid-solid solutions and can be: - substitutional like brass, atoms of zinc replace atoms of the main element, copper, since they are similar size - interstitial like carbon steel, atoms of C fill some spaces between atoms of the main element, iron. Waxes are solids insoluble in water and soluble in nonpolar solvent of biological origin. Thermochemical solution cycle which leads to the formation of a solution: 1. Solute particles separate from each other, ∆H solute > 0 2. Solvent particles separate from each other, ∆H solvent > 0 3. Solute and solvent particles mix and form a solution ∆H < 0 By Hess’ law: ∆H soln = ∆H solute + ∆H solvent + ∆H mix. If it is highly positive, the solute may not dissolve significantly. ∆H solvation = ∆H solvent + ∆H mix. Solvation in water is hydration. ∆H soln = ∆H solute + ∆H hydr. Hydration of an ion is always exothermic. Heats of hydration depend on the ion’s charge density (charge/volume). The higher the charge, the more negative ∆Hhydr is. Down a group the charge stays the same and the size increases, ∆H hydr ↓; across a period the charge is higher and the radius smalle so ∆H hydr ↑. To separate an ionic solute into gaseous ions requires a high ∆H solute. This is the lattice energy and it is highly positive: MX(s) → M+(g) + X-(g) ∆H solute = ∆H lattice. For ionic compounds in H2O ∆H soln = ∆H lattice + ∆H hydr of the ions. NaCl has a small positive ΔHsoln (3.9 kJ/mol) because its lattice energy is only slightly greater than the combined ionic heats of hydration. NaOH has a large negative ΔHsoln (–44.5 kJ/mol) because its lattice energy is much smaller than the combined ionic heats of hydration: if you dissolve NaOH in water, the flask feels hot. The heat of solution is one of the two factors that determine whether solute dissolve. The other is entropy (S). A solution usually has a higher entropy than the pure solvent and solute. Solution formation involves the interplay of two factors: systems change towards a state of lower enthalpy and higher entropy. NaCl in hexane. Separating the nonpolar solvent is easy because the dispersion forces are weak, but separating the solute requires supplying the very large ΔH lattice. Mixing releases little heat because ion–induced dipole forces between Na+ (or Cl−) and hexane are weak → ΔHsoln ≫ 0. A solution does not form because ΔSmix ≪ ΔHsolute. A saturated solution is at equilibrium between the particles that dissolve and those that recrystallize, and contains the maximum amount of dissolved solute at a given T. Some solutes are ore soluble at higher T and this fact is used to cerate supersaturated solutions which eventually cool down into crystals. The effects of T o solubility of solids in H2O in unpredictable, while for gases it is. Gas particles are already separated, ∆H solute +- 0. Because hydration is exothermic, ∆H soln < 0 ad thus gas solubility decreases with rising T .The gases kinetic energy increases, the particles easily overcome the intermolecular forces with water and re-enter the gas phase. Pressure increases gas solubility because more particles collide with the liquid surface more often and more particles enter than leave the solution per unit time. Henry’s law: Sgas = kH * P gas. kH is Henry’s constant in mol/L*atm. Concentration is an intensive property, does not depend on the quantity of mixture. Molarity changes with temperature and sometimes volumes are not additive (500mL solute + 500mL solvent may not give 1000mL of final solution). Molality does not change with T and masses are additive. COLLIGATIVE PROPERTIES The magnitude of colligative properties is strictly related to the ability of solute to conduct a current. Strong electrolytes are soluble salts, strong acids, strong bases; weak electrolytes are weak acid and bases. For nonelectrolytes, 1 mol of compound yields 1 mol of particles when it dissolves; for strong electrolytes, 1 mol of compound yields the amount of ions shown in the formula unit (eg 0.4 M Na2SO4 has 0.8 mol of Na and 0.4 mol of SO4); for weak electrolytes the calculation is different. Non-volatile nonelectrolyte solutions. • Vapour pressure lowering: vapour pressure of solution < vapour pressure of pure solvent. Fewer particles need to vaporize to reach the same entropy of the equilibrium entropy of the solvent and with fewer prticles in the gas phase, the vapour pressure is lower. By Raoult’s law: P sv = X sv * P° sv. X is always < 1 in sn. • Boiling point elevation: a solution boils at a higher T than the pure solvent because the boiling point is the t at which the vapour pressure equals the external pressure. Since the vapour pressure is lower, it takes more heat to equal it and the boiling point is higher. Boiling point elevation is proportional to concentration: ∆Tb = Tb solution - Tb solvent. Kb in °C/m Freezing point depression: a solution freezes at a lower T than the pure solvent. Only solvent vaporizes from a solution and similarly only solvent freezes. The freezing point of a solution is the T at which the vapour pressure equals that of the pure solvent. Since the vapour pressure is always lower, the solution freezes at a lower T. ∆Tf = Tf solvent - Tf solution • Osmotic pressure: a net flow of solvent from the less concentrated side of a semipermeable membrane to the other causes a pressure difference called osmotic pressure. It is the pressure difference at which water is being pushed out of the solution at the same rate it is entering (system at equilibrium). Osmotic pressure is the same pressure that has to be applied to prevent the movement of water to the more concentrated side. • Volatile nonelectrolyte solutions. P solvent = X solvent * P° solvent P solute = X solute * P° solute P total = P solvent + P solute = (X solvent × P° solvent) + (X solute × P° solute) The vapour has a higher mole fraction of the more volatile component. Volatile electrolyte solutions. Van’t Hoff factor i = 1 + α(v +1) α = dissociation degree v = number of ions The factor approaches the ideal value as solutions become more dilute, as the distance between ions increases. Placing a cell in an isotonic solution, one that has the same concentration of particles as the cell fluid, maintains the cell’s normal shape because water enters and leaves the cell at the same rate CHAPTER 16 Reaction rate is the change in concentration of reactants (decrease) and products (increase) as a function of time. It is affected by: 1. Reactants concentration. The more molecules present, the more often they collide. 2. Reactants physical state, when they are in different physical states, contact occurs only at the interface. 3. Temperature of reaction. The most probable speed increases with T, thus, at higher T the frequency of collision is higher. The kinetic energy of collisions is greater too. Collision energy has to be high enough for particles to collide. 4. Use of a catalyst. Since A2 concentration must be lower than A1 concentration, the change in concentration is negative (minus sign to make rate positive). Rate decreases during the course of a reaction. Average rate over the entire period, average rate over a shorter period, last period average rate, instantaneous rate at 35’’ ( the derivative of the curve at a certain time), initial rate (as a reaction proceeds in the forward direction reactants→products, product increases causing the reverse reaction to occur more quickly. To calculate the net rate, we would have to calculate the difference between forward and reverse rates, but for the initial rate products concentrations are negligible and so is the reverse rate). Rate law: k is the rate constant specific for a reaction at a given T and does not change as the reaction proceeds. Its units of measurement depend on the reaction orders m and n, which are not related to the balancing coefficients. Concentration → initial rate → reaction orders → rate constant→rate for any ideal reaction To determine the initial rate: spectrometric methods (color); conductometric methods (conductivity); manometric methods (pressure). Each reaction has an individual order wrt each reactant and an overall order, the sum of individual orders. Reaction: A →products First order: the rate doubles when A doubles, the rate depends on [A]^1; second order: rate quadruples when A doubles, depends on [A]^2; zero order: rate does not change when A doubles, it does not depend on A (=k*[A]^0 = k) The decreases slows down for a first order reaction and it slows even more for a second order one. Reaction: A + B → C + D Rate = k[A]m[B]n. To find m and n we run experiments in which one reactant concentration changes and the other is held constant, measuring the effect on the initial rate in each case. Rate2 / Rate 1 = [X]1 / [X]2. Sometimes the exponent is not so easy to find. In that case we use log10 (Rate2 / Rate1)= m log([A]2/[A]1) and then solve for k = Rate / ([X]1n[X]2m). INTEGRATED RATE LAWS If rate data are not available, we rearrange the integrated rate law into an equation for a straight line. • First order reaction: ln[A]t = -kt + ln[A]0 → if you obtain a straight line when you plot ln [reactant] vs t, the reaction is first order with respect to that reactant. • Second order reaction: 1/[A]t = kt + 1/[A]0 → if you obtain a straight line when you plot 1/[reactant] vs t, the reaction is first order with respect to that reactant. • Zero order reaction: [A]t = -kt + [A]0 → if you obtain a straight line when you plot [reactant] vs. t, the reaction is zero order with respect to that reactant The half-life (t ½ ) of a reaction is the time it takes for the reactant concentration to half its initial valure. A fast reaction (large k) will have a shorter half-life. For a first-order reaction, the time needed to reach half the starting concentration is not dependent from starting concentration. A second-order reaction with a higher initial reactant concentration has a shorter half-life. Each successive half-life is double the preceding one. The half-life of a zero-order reaction is directly proportional to the initial concentration. Collision theory: the number of collisions is depends on the product of the number of reactant particles, not their sum, which is why we multiply the concentrations in rate law to obtain the observed rate. k increases exponentially as T increases. By Arrhenius equation: k = Ae-Ea/RT → T↑ k↑ → Rate↑ Ea is the activation energy: an energy threshold that the colliding particles must exceed in order to react. Ea fwd is the difference between the activated state and the reactants; Ea rev is the energy difference between the activated state and the products. ∆H rxn < 0 → products are at lower energy than reactants, Ea fwd < Ea rev ∆H rxn = Ea (fwd) - Ea (rev) The effect of T on collision energy is a major fact. The fraction of collisions with energy equal to Ea is f = e-Ea/RT A rise in temperature increases the Ek of the reactant particles and enlarges the fraction of collisions with enough energy to exceed Ea. Ea↓ T↑→ f↑ → k↑ → Rate↑ To be effective, a collision must have enough energy and the appropriate molecular orientation. The term A in Arrhenius equation is the frequency factor: the product of the collision frequency Z and an orientation probability factor p, specific for each reaction: A = pZ The more complex the molecular structure, the smaller p. The individual steps that make up a reaction are called elementary steps, characterized by their molecularity, the number of reactant particles in the step. We propose unimolecular or bimolecular reactions as reasonable steps. Reaction order equals molecularity. Only for an elementary step the equation coefficients represent the reaction orders in the rate law. One step is usually much slower than the others: it is the rate-determining step, which becomes the rate law for the overall reaction. A substance formed in one step of the mechanism and used up in a subsequent step during the reaction is a reaction intermediate. The elementary steps must add up to the overall balanced equation; the elementary steps must be reasonable (unimolecular / bimolecular), the mechanism must correlate with the rate law, not the other way around. If the rate-determining step is not the first one, the product of the fast initial step builds up and starts reverting to reactant. With time this step reaches equilibrium. We have to consider the reverse reaction rate too and eliminate the concentration of the intermediate by expressing it in terms of reactants at equilibrium: k1 [A1] [B1] = k -1 [INTERMEDIATE], and then substitute in the rate law for the slow step. We assess the validity of a mechanism with a fast initial step by: 1. Writing rate laws for the fast step (both directions) and for the slow step. 2. Expressing [intermediate] in terms of [reactant] by setting the forward rate law of the reversible step equal to the reverse rate law, and solving for [intermediate]. 3. Substituting the expression for [intermediate] into the rate law for the slow step to obtain the overall rate law. Only reactants involved up to and including the slow (rate-determining) step appear in the overall rate law. Catalysts accelerate the reaction without being consumed. It provides a different reaction mechanism with a lower Ea, which makes the rate higher. Enzymes are protein catalysts. CHAPTER 17 At equilibrium, reactant and product concentration are constant: rate fwd = rate rev. The ratio of the constants creates the equilibrium constant K: • Small K. If a reaction yields little product before reaching equlibrium • Large K. The reaction goes to completion. • Intermediate K. Significant qmounts of reactant and product are present at equilibrium. Law of mass action: a chemical system reaches a state in which a particular ratio of reactant and product concentrations has a constant value. This ratio is called the reaction quotient Q. At equilibrium Q = K. For a particular system and temperature, the same equilibrium state is attained regardless of starting concentrations K is a special value of Q that occurs when the reactant and product concentrations are at equilibrium. Qc, Kc = based on concentration, unitless For all pure solids and liquids, Q = 1, since their concentrations are constant. When the components are in different phases, the equilibrium is heterogeneous. If an overall reaction is the sum of two or more reactions, the overall Q/K are the product of all the Q/K of the steps. At constant T, pressure is directly proportional to molar concentration. Kp is based on partial pressures. To pass from Kc to Kp and vice versa use ∆n (change in amount gas). If the amount of gas does not change, Kp = Kc. ! Do not count solids or liquids when calculating ∆n. • Q < K → the reaction will progress to the right • Q = K → equilibrium • Q > K → the reaction will progress to the left To solve exercises you first have to determine whether the reaction proceeds towards the left or right, to assign values to the x. If a reaction has a small K and large initial reactant concentration, the concentration change (x) can be neglected if [A]init / Kc > 400. Le Chatelier’s principle: when a chemical system at equilibrium is disturbed (Q != K), it reattains equilibrium by undergoing a reaction that reduces the effect of the disturbance (shift to the right or left). The system reacts to consume some of the added substance or produce some of the removed substance to make Qc = Kc again. A change in volume is a change in concentration. A change in pressure due to a change in volume does not alter Kc. Only temperature does. Exothermic reaction: T↑K↓; endothermic: T↑K↑. Van’t Hoff equation: The Haber process for the production of ammonia takes advantage of these facts. It is maximized at high pressure, low temperature and with the continual removal of NH3 produced. The reaction is exothermic and is N2(g) + 3H2(g) → 2 NH3(g). A catalyst is used to increase rate. CHAPTER 18 Water is the product in reactions between strong acids and bases. Acid + base →neutralization ∆H°=-55.9kJ per mol H2O, always. Arrhenius definition: an acid is a substance with H in its formula that dissociates in water to form H3O+; a base is a substance with OH in its formula that dissociates in water to form OH-. Strength is the amount of H3O+ or OH-produced per mole substance dissolved, correlates with electrolyte strength. Strong acids dissociate completely, HA + H2O(l) →H3O+ (aq) + A-(aq), HA ‘disappears’, Kc very large. Weak acids dissociate slightly into ions, Kc is very small. Acid-dissociation constant: The stronger the acid, the higher [H3O+] at equilibrium, the larger Ka. Strong acids: hydrohalic acids HCl, HBr, HI; oxoacids (weak acids) in which the number of O atoms exceeds the number of ionizable protons by two or more; Weak acids: hydrohalic acid HF; acids in which H is not bonded to O or to a halogen, such as HCN and H2S; oxoacids in which the number of O atoms equals or exceeds by one the number of ionizable protons; carboxylic acids (general formula RCOOH). Strong bases: water-soluble compounds containing O2- or OH- ions. The cation is usually a group 1A/2A metal. Weak bases: none is an Arrhenius base, they contain an N atom with a lone electron pair and are ammonia (NH3) and amines (RNH2, R2NH, R3N). Water dissociates very slightly into ions in an equilibrium process known as autoionization. Because the concentration of water (55.5M) stays essentially constant, it is considered as a pure liquid. Ion-product constant for water Kw = [H3O+][OH] = 1.0 * 10^-14 M at 25°C. In pure water [H3O+]=[OH-] = 1 * 10^-7M [H3O+]↑[OH-]↓ and vice versa. If some acid is added, the autoionization process proceeds to the left and the ions react to form water. Both ions are present in all aqueous solutions, we define acidic and basic solutions in terms of relative magnitudes of the two concentrations. Acidic solutions have a higher pOH (lower [OH-]) than basic solutions. Equilibrium constant can be expressed as pK = -log K; pK↓ K↑. FOR STRONG ACIDS AND BASES the eq concentration of hydronium is equal to the initial concentration of a strong acid; the eq concentration of OH- is equal to the initial concentration of a Group 1A hydroxide and twice the one of a Group 2A hydroxide. pH values are obtained with acid-base indicators or with a pH meter, which measures [H3O+] by means of two electrodes. Arrhenius definition didn’t include bases which do not contain OH and water had to be the solvent. Bronsted Lowry acid-base definition: an acid is a proton donor and must contain H in its formula; a base is a proton acceptor and must contain a lone pair of electrons to bind H+. An acid-base reaction is a proton transfer process. Water can thus act both as a base and an acid (amphiprotic). Every acid has a conjugate base and every base has a conjugate acid. For every conjugate pair: • The conjugate base has one fewer H and one more negative charge than the acid • The conjugate acid has one more H and one fewer negative charge than the base A Bronsted-lowry acid-base reaction occurs when an acid and a base react to form their conjugated base and acid: A1 + B2 → B1 + A2. A reaction proceeds in the direction in which a stronger acid and base form a weaker acid and base. With strong acids, H2O wins the competition, while with weak acids the A - base does. A weaker acid has a stronger ocnjugate base: the acid can’t give up its proton very readily because its conjugate base attracts the proton too strongly. A reaction proceeds to the right if an acid reacts with a base lower on the list. Acids with more than one ionizable proton are polyprotic acids and Ka is different for each step, Ka1 >> Ka2 >> Ka3. To determine the acidity of a nonmetal hydride, check the EN of the central atom and the strength of the bond. The strength of oxoacids is determined by the EN of the central nonmetal E and the number of O around E. For oxoacids with the same number of O atoms, the greater EN the easier the proton is released. For oxoacids with different numbers of O, acid strength increases with the number of O atoms. The hydrated cation M(H2O)n+ is a Bronsted-Lowry acid. Hydrolisis: A- + H2O(l) → HA(aq) + OHThe base-dissociation constant Kb: although no base dissociates in the process. The larger Kb, the stronger the base, producing a higher [OH-] at equilibrium. A salt consisting of the anion of a strong acid and the cation of a strong base yields a neutral solution because the ions do not react with water. The ions that do not react with water are: • the anions of the strong hydrohalic acids: Cl−, Br−, I−; • the ions of strong oxoacids, such as NO3 and ClO4 • the Group 1A(1) ions (cations of strong bases); • Ca2+, Sr2+, and Ba2+ in Group 2A(2) (cations of strong base). Salts containing only these anions and cations yield neutral solutions. Two types of salt yield acidic solutions: • A salt consisting of the cation of a weak base and anion of a strong acid. The cation acts as a weak acid. • A salt consisting of small, highly charged metal cation and the anion of a strong acid. “ “ Salts that yield basic solutions are those consisting of the anion of a weak acid and the cation of a strong base. If salt = cation and anion of weak acid and base, strength determined by Ka and Kb. If salt = cation of strong base + anion of polyprotic acid with ionizable ions still attached (these ions are amphiprotic, can either release a proton to water or abstract it from it). Lewis acid-base definition: a base is any species that donates an electron pair to form a bond; an acid is any species that accepts an electron pair to form a bond. It expands the class of acids… the proton itself is one. The product of a Lewis acid-base reaction is an adduct, a single species that contains a new covalent bond. A Lewis acid must have a vacant orbital to accept a lone pair. Electron-deficient Lewis acids are Boron and Aluminium compounds. Molecules with a polar double bond also function as Lewis acids, the pi bond breaks. A metal ion acts as an acid when it dissolves in water. CHAPTER 20 Second law of thermodynamics: all real processes occur spontaneously in the direction that increases the entropy of the universe. ∆S sys = q/T = - ∆S surr ∆S universe > 0. Third law of thermodynamics. A perfect crystal has zero entropy at absolute 0. Entropy is an extensive property. In allotropes, the entropy is higher in the form that allows the atoms more freedom of motion. The standard entropy of reaction ∆S°rxn is the entropy change that occurs when all reactants and products are in their standard states. n↑S↑. m and n are the amounts of products and reactants given by the coefficients on the balanced equation. For a process at constant pressure, ∆S surr = -∆Hsys / T. In an endothermic reaction, the heat absorbed by the system makes ∆S surr < 0. For the process to occur spontaneously, ∆S sys has to be large enough to outweigh the negative change in the surroundings. Gibbs free energy: G = H - TS. The free energy change is a measure of the spontaneity of the process. ∆S univ = ∆S sys - ∆H sys / T → -T∆Suniv = ∆G sys = ∆H sys - T∆S sys Spontaneous process: ∆G < 0 Nonspontaneous process: ∆G > 0 Equilibrium ∆G = 0 ∆G = w max, it is the maximum useful work that can be done by a system during a spontaneous process at T, P = k; it is the minimum work that must be done to a system to make a nonspontaneous process occur “ “. The crossover T which determines whether a process will be spontaneous or not is determined by putting ∆G = 0 If Q and K are very different, the reaction releases (or absorbs) a lot of free energy. If Q and K are nearly the same, the reaction releases (or absorbs) relatively little free energy. When we choose standard states for Q (1 atm or 1M), ∆G° = -RTlnK. In general, ∆G = ∆G° + RT lnQ