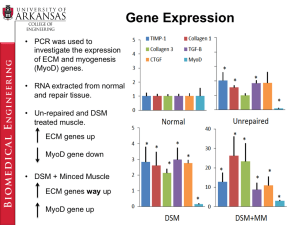
Oncogene (1998) 16, 273 ± 282
1998 Stockton Press All rights reserved 0950 ± 9232/98 $12.00
Functional speci®city of the two retinoic acid receptor RAR and RXR
families in myogenesis
SeÂverine Alric1, Anne FroeschleÂ1, David Piquemal2, Gilles Carnac3 and Anne Bonnieu1
1
Laboratoire de DieÂrenciation Cellulaire et Croissance, Institut National de la Recherche Agronomique (INRA), Place Viala,
34060 Montpellier Cedex 1; 2INSERM U431, Universite Montpellier II Sciences et Techniques du Languedoc, Place EugeÁne
Bataillon, 34095 Montpellier; 3Cell Biology Unit, Centre de Recherches de Biochimie MacromoleÂculaire, CNRS, BP 5051, 34033
Montpellier Cedex, France
In C2 myoblasts, retinoic acid (RA) is an ecient inducer
of both growth arrest and dierentiation. These RA
eects are mediated through at least two classes of
retinoic acid receptors (RARs and RXRs), which belong
to the nuclear receptor superfamily. To determine the
role played by each RAR or RXR family in this model
system, we have analysed the eects of RA in C2
myoblasts expressing a dominant negative RAR (dnRAR)
or a dominant negative RXR (dnRXR). The stable
expression of dnRAR or dnRXR in C2 cells delays the
RA-induced growth arrest and dierentiation, an eect
which is more pronounced in C2-dnRXR myoblasts.
Furthermore, the RA-inducible expression of MyoD gene
is lost in C2-dnRXR but not in C2-dnRAR cells,
indicating that each family of retinoid receptors RAR
and RXR may regulate distinct subsets of RA-responsive
genes. Finally, using C2 cell lines with dierent retinoid
responsiveness, we provided evidence for a link between
the RXR and MyoD families in the process of myogenic
dierentiation. These results illustrate a critical role for
RA-receptors in RA-control of C2 myogenesis and
provide tools for studying the function of RA and its
receptors during vertebrate development.
Keywords: myogenesis; retinoic acid; RAR; RXR;
dominant negative; MyoD
Introduction
Retinoic acid (RA), a natural derivative of vitamin A,
exhibits regulatory properties on growth and differentiation of many vertebrate tissues (De Luca, 1991;
Chambon, 1994; Gudas et al., 1994; Hofman and
Eichele, 1994). The RA actions are believed to be
primarily mediated by retinoic acid receptors (RAR a,
b and g) and retinoid X receptors (RXR a, b and g),
which belong to the nuclear hormone receptor superfamily (Leid et al., 1992a; Chambon, 1994; GigueÁre,
1994; Gronemeyer and Laudet, 1995; Mangelsdorf and
Evans, 1995). The RARs and RXRs display the
modular structure of nuclear receptors with six
distinct regions (denoted A ± F) comprising the region
C or DNA-binding domain (DBD) and the region E,
which mediates ligand binding, ligand dependent
transcriptional activation by AF-2 (Durand et al.,
1994) and dimerization (Forman and Samuels, 1990;
Leid et al., 1992b; Glass, 1994; Chambon, 1996).
Correspondence: Anne Bonnieu
Received 14 March 1997; revised 7 August 1997; accepted 7 August
1997
RARs bind and are activated by all-trans RA and its 9cis isomer (Zelent et al., 1989; Allenby et al., 1993),
while RXRs bind and are activated by 9-cis RA only
(Heyman et al., 1992; Levin et al., 1992; Mangelsdorf
et al., 1992). Transcriptional activation through
retinoic acid response elements is thought to be
mediated by heterodimer formation between RAR
and RXR family members, or by homodimer
formation of RXRs (Leid et al., 1992a; Zhang et al.,
1992; Heery et al., 1993; GigueÁre, 1994; Glass, 1994).
These various RXR-RAR and RXR-RXR combinations within the cell could explain the diversity of RA
eects. Moreover, RXRs not only serve as accessory
factors for RARs in retinoid signaling, but also
function as DNA binding partners for thyroid
hormone receptors (TRs), the vitamin D3 receptor
(VDR), peroxisome proliferator activated receptors
(PPARs) and numerous orphan receptors (Chambon,
1994; GigueÁre, 1994; Glass, 1994; Leblanc and
Stunnenberg, 1995; Mangelsdorf and Evans, 1995).
Thus RXRs are considered to play a central role in the
mediation of several hormonal signals.
The C2 myogenic cell line is a well established model
system for the study of retinoid receptor functions in
the control of proliferation and dierentiation. We
previously reported that RA promotes growth arrest
and myogenesis of C2 myoblasts (Albagli-Curiel et al.,
1993). Furthermore, RA regulates the expression of
some myogenic factors (members of MyoD family)
involved in the activation of the myogenic program
(Albagli-Curiel et al., 1993; Carnac et al., 1993a,b).
Finally, the study of a C2 subclone which failed to
respond to RA has revealed an essential role of RXRa
in mediating RA-induced growth arrest and control of
gene expression in those cells (Froeschle et al., 1996).
However, as RARs and RXRs coexist in C2 cells, it is
dicult to establish a speci®c role for each receptor in
the RA signaling pathway (Carnac et al., 1993a;
Downes et al., 1994). To determine the speci®c role
played by each RAR or RXR family in regulating cell
growth and dierentiation of C2 cells, we have now
compared the eect of RA in C2 myoblasts expressing
a dominant negative RAR (dnRAR) lacking the
ligand-dependent transcriptional activation function
of RARa or a dominant negative RXR (dnRXR)
lacking the DNA binding domain of RXRb. Our
results show that the RA-induced growth arrest and
dierentiation are delayed in C2 cells expressing
dnRAR or dnRXR. Furthermore, this eect is more
pronounced in C2-dnRXR myoblasts. We also
demonstrate that the expression of several RAresponsive genes is impaired in these two types of C2
cell lines. Interestingly, we report that RA-induced
RARs and RXRs functional specificity in C2 cells
S Alric et al
274
expression of the MyoD gene is lost in C2-dnRXR but
not in C2-dnRAR cells, which indicates some
speci®city of RAR and RXR families function in
myogenic cells. Finally, using C2 sublines which dier
in their pattern of retinoid responsiveness, we provided
additional evidence of a link between the RXR and
MyoD families in mediating RA myogenic events.
These results illustrate the contribution of MyoD and
RXR families to muscle cell dierentiation.
pSG5
40
100
–
+
RAREβ-tk-CAT
dnRAR
13
dnRXR
14
42
45
Results
Overexpressed dnRAR or dnRXR acts as dominant
negative inhibitor of RA-induced reporter transcription in
C2 myoblasts
Previous studies of Durand et al. (1992) and Minucci et
al. (1994) respectively described the mutated RARa
lacking the ligand-dependent transcription activation
function (AF-27) and the mutated RXRb lacking the
DNA binding domain (DBD7) which exert a dominant
negative activity in P19 embryonal carcinoma cells.
Before using these mutant receptors (designated in this
work as dnRAR and dnRXR) to block the action of
endogenous retinoic acid receptors in myogenesis, we
®rst tested their dominant negative activity following
transient transfection into C2 myogenic cells. For this
purpose, C2 cells were cotransfected with the expression vectors encoding dnRAR or dnRXR receptors
(pSG5-dnRAR or pSG5-dnRXR) and a reporter
construct containing the classical retinoic-acid response element (RAREb) (de The et al., 1990;
Homan et al., 1990) in front of the thymidine kinase
promoter linked to the chloramphenicol-acetyl transferase gene (tk-CAT) (see Materials and methods)
(Figure 1). As shown in Figure 1, treatment of pSG5transfected C2 cells with RA increased RAREb-tkCAT reporter activity by 2 ± 3-fold. However, when
dnRAR or dnRXR was cotransfected, CAT activity
was strongly inhibited (maximum inhibition =100%).
These results indicate that both types of mutated
RA-receptors inhibited RA-induced activation of
reporter transcription, consistent with the proposed
dominant-negative properties (Durand et al., 1992;
Minucci et al., 1994). This inhibition is likely to be
attributed to impaired function of endogenous RAreceptors through formation of nonfunctional RAR/
RXR heterodimers.
Overexpressed dnRAR or dnRXR inhibits the
RA-induced growth arrest in C2 myoblasts
The eectiveness of the dnRAR or dnRXR in
inhibiting the transcriptional activation of endogenous
RA-receptors in C2 cells prompted us to explore their
ability to disturb the action of endogenous RAreceptors in mediating RA myogenic eects.
A characteristic of RA-treated C2 cells is a marked
slowing of cellular growth as dierentiation progresses.
Here, we have tested whether the expression of
dominant negative RA-receptors alters the normal
RA responsiveness of C2 cells with respect to growth
arrest. C2 cells were cotransfected with the expression
vectors encoding dnRAR or dnRXR receptors together
with pSV2neo as selection marker. Control cells were
RA
–
+
–
+
Figure 1 dnRAR and dnRXR inhibit the transcriptional activity
of retinoic acid receptors in C2 cells. C2 cells were transiently
cotransfected with RAREb-tk-CAT reporter (1 mg) and an
equivalent amount of the respective expression vectors: pSG5
(control), pSG5-dnRAR, pSG5-dnRXR as indicated. Cells were
grown in DMEM/HamF12 supplemented with 10% depleted
FCS. 24 h after transfection, the cells were stimulated (+) or not
(7) with 1076 M RA for an additional 24 h before harvesting and
determination of CAT activity (see Materials and methods). CAT
activities are expressed relative to that in (+) RA pSG5transfected control cells (arbitrary taken as 100). A representative
experiment from three independently performed ones is shown
cotransfected with pSG5 plasmid without insert plus
pSV2neo. Cells in proliferation medium (see Materials
and methods) were selected by G418 alone, or by G418
plus RA (1076 M), during a 2 ± 3 weeks period. At the
end of the culture, colonies were stained with Giemsa
stain and counted (Figure 2). When selected by G418
alone, a large number of colonies (100 ± 150) were
produced by transfection with the dominant-negative
receptors or the control plasmid. However, when
selected by the G418 plus RA, only a few colonies
emerged by transfection with the control plasmid. In
contrast, transfection with dnRAR or dnRXR resulted
in a larger number of colonies surviving the selection
(RA+G418). Note that the number of RA-resistant
cells is greater in dnRXR than in dnRAR transfected
cells (Figure 2). These results were observed reproducibly in three separate experiments. These data
demonstrate that C2 myoblasts expressing dnRAR or
dnRXR fail to arrest cell growth in response to RA. It
is interesting to note that, in absence of RA, mutated
receptors reduced the proliferative capacity of C2 cells,
a result also obtained with C2 cells overexpressing
intact RAR or RXR (Figure 2 and data not shown).
This observation could be explained by the ligandindependent transactivation function (AF-1) contained
within the A/B region of these wild type and mutated
retinoic acid receptors (Nagpal et al., 1992, 1993).
RARs and RXRs functional specificity in C2 cells
S Alric et al
et al., 1996). In contrast, the dominant negative
receptors transfected C2 cell populations exhibited
marked blunting of this dierentiative response to
RA with no more than 10 ± 18% of the cells
dierentiated after exposure to RA. Note that C2dnRXR1 cell dierentiation seems to be twofold more
resistant to RA than C2-dnRAR1 dierentiation.
Similar results were obtained for C2-dnRAR2 and
C2-dnRXR2 cells (data not shown). Thus, the overexpression of dominant negative RA-receptors delays
the RA-induced dierentiation process in C2 cells.
These results demonstrate that in C2 cells expression
of dnRAR or dnRXR leads to inhibition of RAinduced growth arrest.
Overexpressed dnRAR or dnRXR delays the RA-induced
dierentiation in C2 myoblasts
To further investigate the impact of the dominantnegative RA-receptors eects on the RA-induced
dierentiation process, we established stably dnRARor dnRXR- expressing C2 cells by cotransfecting
expression vectors encoding either dnRAR or dnRXR
together with the pSV2neo as described above (see
Materials and methods). Several stably transfected cell
populations were selected and characterized. These cell
populations designated C2-dnRAR and C2-dnRXR
expressed dierent levels of dnRAR or dnRXR
transcripts which were, as expected, undetectable in
C2-neo cells (Figure 3a). It is of note that levels of
dominant negative receptor transcripts in the selected
cell populations were comparable to the level of the
endogenous corresponding receptor.
We then examined the dierentiation response of
these dierent stably transfected C2 cell populations to
RA. Among the four cell populations tested, the RAinduced dierentiation of representative C2-dnRAR1
and C2-dnRXR1 cell populations was evaluated by
immuno¯uorescence analysis. Myoblasts were induced
to dierentiate in low serum medium supplemented
with RA (1076 M) during 3 days (see legend Figure 3b),
then immunostained for troponin T, a late muscle
marker. The control C2-neo cells exhibited a similar
response to RA as detailed previously for the parental
C2 cells with 65% of the cultured cells undergoing
dierentiation after 3 days of RA-treatment (FroeschleÂ
–RA
pSG5
+RA
Overexpressed dnRAR or dnRXR results in impaired
RA-induction of several genes during myogenic
dierentiation
Both C2-dnRAR1 and C2-dnRXR1 cell populations
were investigated in detail to assess their ability to
dierentiate in response to RA, as measured by the
expression of RA-responsive genes. The two dominantnegative C2 cell populations and the C2-neo control
cell line were treated or not with 1076 M RA and the
dierentiation was monitored by Northern blot
analysis (Figure 4). The expression of three genes
known to be regulated by RA in C2 cells, namely
RARb, MyoD and myogenin was compared in the
three stable transfectants.
In various cell types including the C2 cell line,
expression of RARb is induced following RAtreatment (Albagli-Curiel et al., 1993; Carnac et al.,
1993b). Furthermore, the RAREb response element
has been described and characterized in the regulatory
region of RARb gene (de The et al., 1990; Homan et
al., 1990). In C2-neo control cells, RARb mRNA was
undetectable before RA-treatment but was strongly
dnRXR
dnRAR
–RA
+RA
–RA
+RA
Figure 2 dnRAR and dnRXR antagonize the RA-induced growth arrest in C2 cells. C2 cells were co-transfected with pSV2neo
and the indicated plasmids and were selected by G418 in the presence (+RA) or absence (7RA) of 1076 M RA for 2 weeks. Plates
(100 mm in diameter) were ®xed and stained with Giemsa stain. Representative colony formation assay and graph corresponding to
several independent experiments are shown
275
RARs and RXRs functional specificity in C2 cells
S Alric et al
C2-dnRXR1
C2-dnRXR2
C2-neo
C2-dnRAR1
a
C2-dnRAR2
induced by 6 h of treatment (Figure 4). In contrast, in
both dominant-negative transfectants, RARb mRNA
was not RA-inducible, consistent with the transient
transfection data in parental C2 cells (see Figure 1).
These results indicate that the RA-induction of RARb
gene is abolished by the expression of mutant RAR or
RXR receptors consistent with the report of Durand et
al. (1992) that RA transcriptional activation of RARb
gene is mediated by RAR-RXR heterodimers.
We next compared the RA-regulation of MyoD and
myogenin gene expression over a time course period of
RA treatment for C2-neo, C2-dnRAR1 and C2dnRXR1 cells. In C2 cells, MyoD is constitutively
expressed at the myoblast stage while myogenin only
appears at dierentiated stage (Montarras et al., 1989).
It has been documented that both MyoD and
myogenin genes are upregulated in response to RA in
C2 cells (Albagli-Curiel et al., 1993). As shown in
Figure 4, myogenin mRNA levels were readily
detectable at 48 h in absence of RA-treatment in the
three cell lines. However they were lowered in
dominant-negative cells compared with the C2-neo
cells, indicating a delay in the spontaneous differentiation process. In presence of RA, an earlier induction of
myogenin occured in C2-neo cells (at 24 h) while this
induction was reduced and delayed in dominant-
C2-neo
276
—
— RARα
— RXRβ
—
— dnRARα
— dnRXRβ
S26
S26
b
RA-treated cells
Differentiation
Troponin T
Hoechst
(percentage)
C2-neo
65%
C2-dnRAR1
18%
C2-dnRXR1
10%
Figure 3 Analysis of stable C2 transfectants expressing dnRAR
or dnRXR. (a) Determination of dnRAR and dnRXR mRNA
levels in dnRAR- or dnRXR-transfected C2 cells. C2 cell
populations co-transfected with pSV2neo+pSG5-dnRAR (C2dnRAR1 and 2), with pSV2neo+pSG5-dnRXR (C2-dnRXR1
and 2) or with pSV2neo+pSG5 (C2-neo) were cultured for 48 h
in proliferation medium (see Materials and methods). Total RNA
(20 mg/lane) was analysed. Hybridization was carried out using
RARa- or RXRb-speci®c cDNA probes. Comparison of RNA
loading is shown by the hybridization of S26 cDNA probe to the
corresponding ®lters. (b) C2-dnRAR and C2-dnRXR cell
populations are resistant to RA-induced myogenic differentiation. C2-neo, C2-dnRAR1 and C2-dnRXR1 cell populations were
induced to dierentiate in DMEM with 2% depleted FCS and alltrans RA (1076 M) during 3 days, then immunostained with
troponin T monoclonal antibody (Sigma) as indicated. In all
cases, nuclei were stained with Hoechst B2883 dye
negative C2 cells (Figure 4). Concerning MyoD,
comparable basal level gene expression was observed
in the three cell lines prior to RA-treatment. After
addition of RA, the levels of MyoD induction are
similar both in C2 control and C2-dnRAR1 cells. In
contrast, in C2-dnRXR1 cells, RA failed to increase
MyoD gene expression. These data were reproducibly
observed with multiple preparations of RNA from
dierent
dominant-negative
transfectants
(C2dnRAR2, C2-dnRXR2).
Thus, although the C2-dnRAR1 and C2-dnRXR1
cell populations behave in a similar fashion with
respect to the RA-regulation of the RARb gene and
the myogenin gene, they yet dier in RA-mediated
regulation of MyoD gene. These results suggest that
each RAR and RXR family may regulate dierent
subsets of RA-responsive genes.
Evidence for a link between RXRa and MyoD
The above results led us to ask whether RXR and
MyoD gene expressions are linked during the RAinduced myogenesis. To address this question, we
exploited a C2 variant cell line, previously named
`Inducible', isolated and characterized by Pinset et al.
(1988). In opposition to parental C2 myoblasts,
inducible myoblasts are not autonomous for differentiation and require insulin or IGFs to undergo
terminal dierentiation (Pinset et al., 1988). These two
C2 cell lines dier also in their MyoD gene expression.
When compared to progenitor C2 cells, inducible cells
do not express the MyoD gene at the myoblast stage
(Montarras et al., 1989). However, overexpression of
MyoD in this variant restores the C2 parental
phenotype (Montarras et al., 1991). Recently, we
reported that, unlike parental C2 cells, inducible cells,
renamed C2-R, are resistant to the RA myogenic
eects and lack RXRa gene expression at the myoblast
stage (Froeschle et al., 1996). Taken together, these
observations led us to propose that RXRa and MyoD
are linked and may be required for the RA-induction
of myogenesis in C2 cell lines.
To address this issue, we compared the expression of
RXRa in C2-R and C2-R/MyoD cells (a previously
described C2-R cell line which constitutively express
MyoD) (Montarras et al., 1991) treated or not with
RA (1076 M) (Figure 5a). Interestingly, Northern blot
analysis revealed the presence of RXRa transcripts
only in C2-R/MyoD cells. Furthermore, in the same
cells, RA induces myogenin gene expression, indicating
a C2 cell phenotype responsive to RA. We next
examined the contribution of MyoD in mediating
RA-induced dierentiation events. For this purpose,
C2-R and C2-R/MyoD cells were plated in proliferation medium (see Materials and methods), treated with
RA (1076 M) during 4 days then proliferation of the
cells was evaluated using [3H] thymidine incorporation
experiments. As shown in Figure 5b, RA has no eect
on the proliferation of C2-R cells while it inhibits the
growth of C2-R/MyoD cells. To determine whether
these RA-treated cells display a dierentiated phenotype, we carried out, under the same experimental
conditions, immunu¯uorescence analysis using an antitroponin T antibody (Figure 5c). After 4 days of RAtreatment, C2-R cells failed to depict any positive
staining for troponin T. In contrast, C2-R/MyoD cells
RARs and RXRs functional specificity in C2 cells
S Alric et al
277
a
b
Control
RA
0 6 12 24 48 6 12 24 48 Hours
RARβ
Myogenin
C2-neo
MyoD
S26
RARβ
Myogenin
C2-dnRAR1
MyoD
S26
RARβ
Myogenin
C2-dnRXR1
MyoD
S26
Figure 4 Eects of RA on the expression of several RA-responsive genes during the dierentiation of C2-dnRAR and C2-dnRXR
cells. (a) C2-neo, C2-dnRAR1 and C2-dnRXR1 cell populations were cultured in proliferation medium (see Materials and methods) in
the absence or presence of 1076 M RA. Total RNA (20 mg/lane) was harvested at the indicated times and subjected to Northern blot
analysis using speci®c cDNA probes for RARb, myogenin, MyoD or S26. (b) The data from panel a were quanti®ed by densitometry
and expressed relative to S26 mRNA levels which serve as control of RNA loading. This experiment was performed twice
expressed troponin T. Consistent with results obtained
for their proliferation, C2-R/MyoD cell dierentiation
is sensitive to RA. Thus, overexpression of MyoD
restored RA-induced dierentiation of C2-R cells.
In conclusion, these results establish that forced
expression of MyoD in C2-R myoblasts, which express
neither MyoD nor RXRa and do not dierentiate
upon RA treatment (Figure 5 and Froeschle et al.,
1996), restores both expression of the RXRa gene and
RA-induced dierentiation as in parental C2 cells.
Discussion
We are interested in exploring in detail the mechanisms
by which RARs and RXRs might orchestrate the
balance between growth and dierentiation in myogenesis. RARs and RXRs mediate most, if not all, of RA
actions. C2 cells contain both RARs and RXRs, and
thus the eects of RA on cellular dierentiation and
proliferation may re¯ect activation of either RARs or
RXRs or both. One way to examine the role of RARs
and RXRs in myogenesis is to utilize dominant
negative receptor constructs that would suppress
normal RARs, RXRs function in myoblasts and then
determine whether the expression of these constructs
would alter the RA-control of growth and differentiation of these myoblasts. In this report, we have
analysed the eects of RA in permanent C2 myoblast
lines expressing a dominant negative RAR (dnRAR)
lacking the ligand-dependent transcriptional activation
function of RARa or a dominant negative RXR
(dnRXR) lacking the DNA binding domain of
RXRb. We show here that expression of these
mutated receptors in C2 cells prevents several aspects
of the RA-associated dierentiation pathway observed
in C2 cells. Dominant negative receptors expression
results in global alteration of RA-regulated growth and
dierentiation in C2 cells, suggesting that retinoic acid
receptors play a crucial role in the myogenic program.
Our results reveal that the lack of RA action is always
more substantial in C2-dnRXR than in C2-dnRAR
cells. At the molecular level, the RA-impaired
dierentiation is re¯ected by altered expression of the
transcripts of several RA-responsive genes, including
RARb, myogenin and MyoD. However, these mutated
RA-receptors exhibit some functional speci®city, as the
RA-inducible expression of the MyoD gene is inhibited
in C2-dnRXR but not in C2-dnRAR cells. Thus, the
present results strongly suggest that both RARs and
RXRs are involved in RA-mediated dierentiation
events; however they also suggest that the RXR family
may play a major role in transducing the RA signal
triggering C2 cell dierentiation.
The dominant negative activities described here are
reminiscent of previously reported inhibitory eects
seen in a number of normal or transformed cells from
dierent tissues expressing truncated or otherwise
mutated RA-receptors, or decreased levels of these
receptors. Examples include a translocation breakpoint
in RARa at the genetic lesion of acute promyelocytic
leukemia (de The et al., 1991; Kakizuka et al., 1991;
Kastner et al., 1992; Grignani et al., 1993, 1995; Perez
et al., 1993; Rousselot et al., 1994), expression of
RARs and RXRs functional specificity in C2 cells
S Alric et al
cases, proliferation characteristics and/or retinoidinduced dierentiation are altered. Therefore, the
alteration of RA eects on the growth and differentiation of normal, embryonic and malignant cells is often
associated with ± or even caused by ± an abrogation of
RA-receptors expression and/or function.
RXRs are thought to exert pleiotropic functions,
since besides their capacity to homodimerize, they are
able to heterodimerize with multiple nuclear receptors
(Chambon, 1994; GigueÁre, 1994; Glass, 1994; Leblanc
and Stunnenberg, 1995; Mangelsdorf and Evans, 1995).
dominant negative RARa mutants in retinoid-resistant
embryonic carcinoma lines (Pratt et al., 1990; Kruyt et
al., 1992), in RA-resistant HL-60 cells (Collins et al.,
1990; Robertson et al., 1992), expression of dominant
negative RXRb mutant in P19 embryonal carcinoma
cells (Minucci et al., 1994), absence of RXRa
expression in RA-resistant myogenic C2 cells
(Froeschle et al., 1996) and F9 embryonal carcinoma
cells bearing single and compound RAR and RXR
mutations (Boylan et al., 1993, 1995; Cliord et al.,
1996; Chiba et al., 1997). Interestingly, in all these
a
C2-R
–
+
C2-R
/MyoD
–
+ RA
RXRα
Myogenin
MyoD
S26
b
C2-R
C2-R + RA
C2-R/MyoD
C2-R/MyoD + RA
100
Proliferation (%)
100
Proliferation (%)
278
60
20
60
20
0
0
1
2
3
c
4
Days
1
2
3
4
Days
RA-treated Cells
C2-R
C2-R/MyoD
Troponin T
Hoechst
Figure 5 MyoD and RXRa are required partners for RA-induced growth arrest and dierentiation of C2-R cells. (a) Consequence
of the forced expression of MyoD in C2-R cells. C2-R and C2-R/MyoD cells, cultured in proliferation medium were treated for
15 h with 1076 M RA, harvested and their RNA analysed by Northern blots. Poly(A)+ RNA corresponding to 200 mg total cellular
RNA were analysed. Filters were probed sequentially for RXRa, myogenin, MyoD and S26 as indicated. Homogeneity in RNA
loading is con®rmed by S26 hybridization. C2-R and C2-R/MyoD myoblasts were maintained in proliferative conditions (see
Materials and methods) then treated daily for 4 days with 1076 M RA. (b) Graphic representation of the proliferative capacity of
the C2-R, and C2-R/MyoD cells evaluated by [3H]thymidine incorporation in the presence or absence of RA as indicated.
Proliferation index is expressed as a percentage; the maximum of [3H]thymidine incorporation being arbitrary taken as 100% for
each of the two cell lines. (c) Immuno¯uorescence analysis was carried out on day 4. The pictures correspond to RA-treated C2-R
and C2-R/MyoD cells (+RA), ®xed and stained for the expression of troponin T as indicated. In all cases, nuclei were stained with
Hoechst B2883 dye. These experiments were carried out three times and yielded similar results
RARs and RXRs functional specificity in C2 cells
S Alric et al
Thereby, the dierent responsiveness to RA of the two
types of dominant-negative C2 cell transfectants may
involve other RXR partners. Possible candidates for
such a partnership could be the orphan receptors, LXR
(Willy et al., 1995) and NGF1-B-Nur77 or the closely
related receptor NURR1 (Forman et al., 1995;
Perlmann and Jansson, 1995), which can heterodimerize with RXRs and confer RXR-speci®c ligand
activation of transcription. It is thus conceivable that
the RA treatment of C2 cells may activate such RXRdependent pathway. However this issue requires a
thorough study concerning the role of these orphan
receptors in muscle cell dierentiation. Whatever
contributions by RXR-dependent pathways may exist,
it is clear from the present study that the lack of RA
action on myogenesis is much more substantial in C2dnRXR than C2-dnRAR cells.
It is also possible that the dierent retinoid
responsiveness between the two dominant negative
receptor cell lines might rely on the RA regulation of
their target genes expression. As shown in Figure 4,
these two cell lines do dier in the regulation of MyoD
expression. Indeed, only C2-dnRXR cells were
defective in RA-induced mRNA expression of MyoD.
Hence, this could underlie some of the dierences in
myogenic response to RA among these two types of
cell lines. This possibility is supported by numerous
observations which indicate a positive correlation
between the level of MyoD gene expression and the
ability of myogenic cell lines to terminally dierentiate
(Pinset et al., 1988; Konieczny et al., 1989; Lassar et
al., 1989; Montarras et al., 1989, 1991; Tapscott et al.,
1989; Froeschle et al., 1996). Thus, it appears that the
impaired RA-induction of MyoD in C2-dnRXR cells
could lead to a more severe C2 phenotype defective in
RA-regulated growth and dierentiation. The molecular basis for this distinct response of MyoD to dnRAR
and dnRXR is unknown but could be related to the
presence of RXRE in this gene. Alternatively or
concomitantly, the activity of MyoD protein could be
dierentially modulated in these transfectants. In this
respect, we have recently shown that RA-receptors
interact with MyoD and upregulate its activity on
skeletal
muscle
program.
Interestingly,
a
dnRXRDDBD which did not interact with MyoD
failed to potentiate MyoD transcriptional activity (our
unpublished data).
MyoD has been implicated as a master regulatory
gene in the process of muscle dierentiation: it is
sucient to induce withdrawal from cell cycle and
expression of muscle dierentiation markers (Crescenzi
et al., 1989; Sorrentino et al., 1990; Weintraub et al.,
1991). These two myogenesis-related events are also
regulated by retinoic acid suggesting that RA-receptors
and MyoD might act in the same regulatory pathway
governing the RA-induced myogenesis. Several lines of
evidence support the notion that cooperation between
the MyoD and RXR families is likely to play an
important role in the process of myoblast differentiation: (i) We have shown here that only dnRXR is able
to inhibit RA induction of MyoD expression and
myogenesis, suggesting a speci®c role of RXR family in
these events; (ii) The RXR receptor has already been
suggested to be an integral component of the myogenic
dierentiation pathway since we found that in RAresistant C2 cells (C2-R), susceptibility to RA-induced
growth arrest could be restored by transfection of a
RXRa expression vector (Froeschle et al., 1996).
However, whether RXRa could induce RA-growth
arrest, it could not activate the fully dierentiated
program suggesting a lack of one or more mediators of
dierentiation inducing activity of retinoic acid in this
system. The myogenic factor MyoD which is absent at
the myoblast stage in this cell line may be such a
factor; (iii) A key ®nding in the present study has been
the observation that overexpression of MyoD in this
C2-resistant cell line restores the fully RA myogenic
actions (i.e. the dierentiation and anti-proliferative
eects of retinoids) but also induces the expression of
RXRa gene. This suggests a special relationship betwen
these two factors. There are several possible explanations, which are not mutually exclusive for this
cooperative interaction. One is that there is a direct
protein-protein interaction between RXRa and MyoD
that increases the activity of either or both factors.
This appears indeed to be the case since we recently
established that members of the MyoD family and
retinoic acid receptors are partners in retinoic acidinduced myogenesis and that this cooperativity is
mediated by direct protein-protein interaction between
(the DBD of) these heterologous classes of transcription factors. Another possibility is that, as a
transcription factor, MyoD participates directly or
indirectly in the control of RXRa gene expression.
However, this issue must await the identi®cation of
RXRa regulatory sequences. Taken together, these
data strongly suggest that coexpression of MyoD and
RXRa could lead to RA-induced muscle differentiation.
It should be noted that growing evidences in the
literature underscore the role of RXRs in the
mediation of the developmental retinoid signal in
vivo. Indeed, the recent phenotypic characterization of
all combinations of RXR (either a, b or g)/RAR (either
a, b or g) compound mutants has provided genetic
evidence supporting the proposal that RXR-RAR
heterodimers act as functional units transducing the
retinoid signal in vivo and furthermore has indicated
that RXRa is the functionally predominant RXR in
vivo (Kastner et al., 1994, 1996a). In addition, it has
been shown that RXRa null mutants are resistant to
RA-induced limb defects (Sucov et al., 1995). Taken
together, these data reveal the importance of RXRa as
an integral component of the RA signalling cascade in
vivo. However, the question about the role of RXRs
and in particular RXRa in the transduction of the RA
signal during muscle development remains.
Retinoic acid is an important signaling molecule in
embryonic development (Tabin, 1991) and several
evidences suggest a role for RA in the determination
of the muscle lineage (Chen and Solursh, 1991; Sive
and Cheng, 1991). This is supported by the presence of
retinoids in myogenic precursor cells (Wagner et al.,
1990, 1992; Chen et al., 1992) and RA-receptors in
muscle tissue (Mangelsdorf et al., 1992; Dolle et al.,
1994; GigueÁre, 1994). However, the knock-outs of
RARs and RXRs in the mouse to date did not
compromise skeletal muscle development (Kastner et
al., 1994, 1996b; Sucov et al., 1994; Lohnes et al., 1995;
Krezel et al., 1996) probably due to a large degree of
functional redundancy among these receptors which
prevented a de®nitive assignment of their physiological
279
RARs and RXRs functional specificity in C2 cells
S Alric et al
280
functions in the animal. To disable completely the
retinoid pathway in skeletal muscle, it will be necessary
to generate multiple knock-outs of RARs and RXRs.
Alternatively, the targeted expression of a dominant
negative retinoic acid-receptor in transgenic mice
allows to investigate the stage- and organ-speci®c
roles of retinoids in mammalian development. This
latter approach was used with success to evaluate the
role of retinoids in epidermal development (Imakado et
al., 1995; Saitou et al., 1995). In this study we have
described the eects of two dominant negative RAreceptors which are capable of inhibiting wild type
receptor function in C2 myogenic cells. Targeted
expression of these dominant negative mutants in the
muscle of transgenic mice will be helpful to evaluate
the function of retinoids and RA-receptors during cell
dierentiation and embryonic development of this
tissue.
Materials and methods
Cell culture products
Dulbecco's modi®ed Eagle's medium (DMEM), nutrient
mixture F-12 (Ham), Fetal Calf Serum (FCS) and
Geneticin (G-418) were purchases from Gibco-BRL. Alltrans retinoic acid (RA) was obtained from Sigma. Alltrans RA was diluted in dimethyl sulfoxide (DMSO)
whereas G418 was diluted in PBS.
Cell culture conditions
Permissive C2.7, inducible myoblasts (designated C2-R in
this study) and inducible cells stably transfected with the
mouse MyoD cDNA (C2-R/MyoD) have been previously
described (Pinset et al., 1988; Montarras et al., 1991).
Proliferating myoblasts were routinely maintained in
proliferation medium (1 : 1 mixture of DMEM and
HamF-12 supplemented with 10% FCS) and incubated at
378C under 5% CO2. Before any treatment with all-trans
RA, cells were grown for about 7 days in proliferation
medium containing 10% hormone-depleted FCS. Depleted
serum was obtained using the resin procedure of Samuels
et al. (1979). All RA-treatments were performed 48 h after
plating at a density of 26103 cells/cm2. For dierentiation,
cells were plated at a density of 46103 cells/cm2, grown for
3 days in proliferation medium containing 10% hormonedepleted FCS and then transferred into dierentiation
medium (DMEM supplemented with 2% hormonedepleted FCS). RA or solvent (0.1% DMSO) were added
simultaneously to the medium change.
Stable transfection of C2 cells with dnRAR or dnRXR
expressing vectors
C2 cells were co-transfected using DOTAP reagent
(Boehringer) as described by the supplier with DNA from
pSG5 vectors containing or lacking the murine dnRAR or
dnRXR coding sequence (Durand et al., 1992; Minucci et
al., 1994) and pSV2neo DNA carrying the neomycin
marker. For each transfection, 10 mg of the dnRAR or
dnRXR expression vector and 500 ng of the pSV2neo
(molar ratio was 20 : 1) were used. The transfected cells
were selected in the presence of 1 mg/ml G-418 (Geneticin,
Gibco-BRL) for 10 ± 15 days. Individual colonies (10 ± 12)
were isolated then passaged into stable cell lines.
Expression of dnRAR and dnRXR in these cell populations was analysed by Northern blotting. C2 cells
transfected by pSV2neo and pSG5 (C2-neo cells) were
also selected and used as control cells.
Colony formation assay
C2 cells (105 cells) seeded in a 100-mm-diameter dish were
cotransfected with 200 ng of pSV2neo and 10 mg of the
expression vector (either pSG5-dnRAR, pSG5-dnRXR, or
pSG5 without insert) for 24 h and were then immediately
exposed to G418 (1 mg/ml). After 24 ± 36 h, all-trans RA
(1 mM) was added to half of the culture. Cells were fed
with fresh G418 and RA every 2 ± 3 days for up to 2 weeks,
until macroscopic colonies developed. Plates were ®xed
with methanol and stained with Giemsa stain (Sigma) and
the number of colonies was counted.
RNA extraction and Northern blot analysis
Total RNA was prepared using guanidinium thyocyanate as
previously described (Chomczynski et al., 1987). When
needed, poly(A)+ RNA was puri®ed on oligo(dT) cellulose
(Pharmacia). For Northern blot analysis, total RNA (20 mg)
and poly(A)+ RNA (corresponding to 200 mg total RNA)
were run on a 2 M formaldehyde-containing 1% agarose gel,
transferred and bound to nylon membranes (Hybond,
Amersham) as described by the supplier. Filters were
prehybridized and hybridized in a mixture containing 50%
formamide, 5 mM NaPO4, 0.75 M NaCl, 1 mM EDTA, 0.5%
sodium dodecyl sulfate (SDS), 0.4 mg/ml denaturated DNA
salmon sperm, 106Denhardt solution, 1% dextran sulfate
and the appropriated probe (106 c.p.m./ml) at 428C and
washed twice (30 min each) in 0.26 standard saline citrate
(SSC)/0.1% SDS at 658C. Filters were hybridized using the
following cDNA probes labeled by random priming: mouse
RAR a, b and RXR a, b (Zelent et al., 1989), mouse MyoD
(Davis et al., 1987), mouse myogenin (Edmondson et al.,
1989) and hamster ribosomal S26 protein (Vincent et al.,
1993). Radioactivity on the nylon membranes was
determined on a PhosphorImager analyser.
Transient transfections and CAT assays
C2 cells were plated at a density of 76103 cells/cm2 (in
60 mm plate) in proliferation medium supplemented with
10% hormone-depleted serum. After 16 h, transfections of
plasmid DNA were performed using DOTAP reagent
(Boehringer) as described by the supplier. Brie¯y, cells
were transfected with 1 mg of reporter construct (RAREbtk-CAT), 2 mg of b-galactosidase expression vector
(PCH110-Pharmacia) and 1 mg of receptor expression
vectors (either pSG5-dnRAR or pSG5-dnRXR) or the
parental expression vector pSG5 (see Figure legend).
Transfection mixtures were always adjusted to 4 mg of
DNA per plate. Cells were exposed to the DNA for 8 h
then refed with DMEM/HamF-12 medium supplemented
with 10% depleted FCS. RA treatment (1076 M) was
performed 24 h after transfection. Determination of CAT
activity was performed as previously described (Pfahl et al.,
1990). The b-galactosidase activity was measured as
previously described (Nilsen et al., 1983) to normalize for
transfection eciency.
The reporter construct RAREb-tk-CAT has been previously described by Durand et al. (1992).
Immuno¯uorescence
Cells were grown for 96 h in proliferation medium, then
shifted in dierentiation medium and treated or not with
RA (see above). After 72 h, cells were ®xed for 5 min in
3.7% (wt/vol) formaldehyde in PBS followed by a 30 s
extraction in 7208C acetone. Expression of troponin T
was assayed using 1 h incubation of cells with a mouse
monoclonal antibody against troponin T diluted 1 : 100
(Sigma). Cells were stained with Hoechst B2883 dye and
mounted in Airvol 205 (15% Airvol 205, Air Products,
Utrecht, the Netherlands, 33% glycerol, 0.1% NaN 3 in
RARs and RXRs functional specificity in C2 cells
S Alric et al
PBS, pH7). Stained cells were observed under microscope
(Axiophot, Carl Zeiss, Inc., Thornwood, NY) using a
planapochromat 406 objective. Fluorescent images were
recorded onto TMAX 400 ®lm (Eastman Kodak Co.,
Rochester, NY).
3
[ H]thymidine incorporation assays
To assay the eect of RA on proliferation, cells were
grown in proliferation medium. The ®rst RA-treatment
(1076 M) was performed a few hours after plating and then
RA was daily pulsed into medium. At daily intervals (17 h
after hormonal treatment), transfected cells were pulsed for
8 h with 2 mCi/ml [3H]thymidine (ICN, France, speci®c
activity 5 Ci/mmol). At the end of labelling period,
transfected cells were rinsed three times with ice-cold PBS
followed by the addition of 2 ml of 5% ice-cold
trichloroacetic acid (TCA), in which the cells were
maintained 10 min. Cells were then rinsed three times
with ethanol 90% and dissolved in 1 ml of 0.1 N NaOH at
378C for 1 h. NaOH suspensions were transferred into
scintillation vials. Radioactivity was measured using 10 ml
liquid scintillation PCS II (Amersham, France).
Acknowledgements
We thank Drs P Vigneron and F Bacou for their continued
interest and support of this work. We would like to thank
Drs C Pinset and D Montarras for proving us with C2-R/
MyoD cell line and Drs P Chambon, P Kastner, O
Minucci, Ph Fort for the various plasmids used in this
work. We are also indebted to Drs H Bernardi, M
Vandromme, P Chuchana, C Bisbal and F Aurade for
fruitful discussions and critical reading of the manuscript.
This work was supported by funds from the Association
FrancËaise contre les Myopathies (AFM), the Association
pour la Recherche sur le Cancer (ARC), the Ligue
Nationale Contre le Cancer, and the Institut National de
la Recherche Agronomique (INRA). SA is the recipient of
a doctoral fellowship from the Ligue Nationale Contre le
Cancer.
References
Albagli-Curiel O, Carnac G, Vandromme M, Vincent S,
CreÂpieux P and Bonnieu A. (1993). Dierentiation, 52,
201 ± 210.
Allenby G, Bocquel MT, Saunders M, Kazmer S, Speck J,
Rosenberger M, Lovey A, Kastner P, Grippo JF,
Chambon P and Levin AA. (1993). Proc. Natl. Acad.
Sci. USA, 90, 30 ± 34.
Boylan JF, Lohnes D, Taneja R, Chambon P and Gudas LJ.
(1993). Proc. Natl. Acad. Sci. USA, 90, 9601 ± 9605.
Boylan JF, Lufkin T, Achkar CC, Taneja R, Chambon P and
Gudas LJ. (1995). Mol. Cell. Biol., 15, 843 ± 851.
Carnac G, Albagli-Curiel O, Desclozeaux M, Vandromme
M, Glineur C, BeÁgue A, Laudet V and Bonnieu A. (1993a).
Oncogene, 8, 3103 ± 3110.
Carnac G, Albagli-Curiel O, Levin A and Bonnieu A.
(1993b). Endocrinology, 133, 2171 ± 2176.
Chambon P. (1994). Sem. in Cell Biol., 5, 115 ± 125.
Chambon P. (1996). Faseb J., 10, 940 ± 954.
Chen Y and Solursh M. (1991). Roux's Arch. Dev. Biol., 200,
162 ± 171.
Chen Y, Huang L, Russo AF and Solursh M. (1992). Proc.
Natl. Acad. Sci. USA, 89, 10056 ± 10059.
Chiba H, Cliord J, Metzger D and Chambon P. (1997).
Mol. Cell. Biol., 17, 3013 ± 3020.
Chomczynski P and Sacchi N. (1987). Anal. Biochem., 162,
156 ± 159.
Cliord J, Chiba H, Sobieszczuk D, Metzger D and
Chambon P. (1996). EMBO J., 15, 4142 ± 4155.
Collins SJ, Robertson KA and Mueller L. (1990). Mol. Cell.
Biol., 10, 2154 ± 2163.
Crescenzi M, Felming TP, Lassar AB, Weintraub H and
Aaronson SA. (1989). Proc. Natl. Acad. Sci. USA, 87,
8442 ± 8446.
Davis RL, Weintraub H and Lassar AB. (1987). Cell, 51,
987 ± 1000.
De Luca LM. (1991). FASEB J., 5, 2924 ± 2933.
De The H, Vivanco-Ruiz MM, Tiollais P, Stunnenberg H
and Dejean A. (1990). Nature, 343, 177 ± 180.
De TheÂ, Lavauc C, Marchio A, Chomienne C, Degos L and
Dejean A. (1991). Cell, 66, 675 ± 684.
Dolle P, Fraulob V, Kastner P and Chambon P. (1994).
Mech. Dev., 45, 91 ± 104.
Downes M, Mynett-Johnson L and Muscat GEO. (1994).
Endocrinology, 334, 2658 ± 2261.
Durand B, Saunders M, Leroy P, Leid M and Chambon P.
(1992). Cell, 71, 73 ± 85.
Durand B, Saunders M, Gaudon C, Roy B, Losson R and
Chambon P. (1994). EMBO J., 13, 5370 ± 5382.
Edmondson DG and Olson EN. (1989). Genes and Dev., 3,
628 ± 640.
Forman BM and Samuels HH. (1990). Mol. Endocrinol., 4,
1293 ± 1301.
Forman BM, Umesono K, Chen J and Evans R. (1995). Cell,
81, 541 ± 550.
Froeschle A, Carnac G, Alric S, Montarras D, Pinset C,
Rochette-Egly C and Bonnieu A. (1996). Oncogene, 12,
411 ± 421.
GigueÁre V. (1994). Endocrine Review, 15, 61 ± 79.
Glass CK. (1994). Endocrine Review, 15, 391 ± 407.
Grignani F, Ferrucci PF, Testa U, Talamo G, Fagioli M,
Alcalay M, Mencarelli A, Grignani F, Mencarelli A,
Grignani F, Peschle C, Nicoletti I and Pelicci PG. (1993).
Cell, 74, 423 ± 431.
Grignani F, Testa U, Fagioli M, Barberi T, Masciulli R,
Mariani G, Peschle C and Pelicci PG. (1995). Cancer Res.,
55, 440 ± 443.
Gronemeyer H and Laudet V. (1995). Protein Pro®le, 2,
1173 ± 1236.
Gudas LJ, Sporn MB and Roberts AB. (1994). The Retinoids,
Sporn MB, Roberts AB and Goodman DS. (eds.). 2nd
Edn., Raven Press, Ltd: New York, pp. 443 ± 520.
Heery D, Pierrat B, Gronemeyer H, Chambon P and Losson
R. (1993). Proc. Natl. Acad. Sci. USA, 90, 4281 ± 4285.
Heyman RA, Mangelsdorf DJ, Dyck JA, Stein RB, Eichele
G and Evans RM. (1992). Cell, 68, 397 ± 406.
Homan B, Lehman JM, Zhang XK, Hermann T, Husmann
M, Graupner G and Pfahl M. (1990). Mol. Endocrinol., 4,
1727 ± 1736.
Hofman C and Eichele G. (1994). The Retinoids, Sporn MB,
Roberts AB and Goodman DS. (eds.). 2nd Edn., Raven
Press, Ltd: New York, pp. 387 ± 441.
Imakado S, Bickenbach JR, Bundman DS, Rothnagel JA,
Attar PS, Wang X-J, Walczak VR, Wisniewski S, Pote J,
Gordon JS, Heyman RA, Evans RM and Roop DR.
(1995). Genes and Dev., 9, 317 ± 329.
Kakizuka A, Miller WH, Umesono K, Warrel RP, Frankel
SR, Murty VS, Dmitrovsky E and Evans RM. (1991). Cell,
66, 663 ± 674.
Kastner P, Perez A, Lutz Y, Rochette-Egly C, Gaub M-P,
Durand B, Lanotte M, Berger R and Chambon P. (1992).
EMBO J., 11, 629 ± 642.
281
RARs and RXRs functional specificity in C2 cells
S Alric et al
282
Kastner P, Grondona JM, Mark M, Gansmuller A, LeMeur
M, Decimo D, Vonesh JL, Dolle P and Chambon P.
(1994). Cell, 78, 987 ± 1003.
Kastner P, Mark M, Ghyselinck N, Krezel W, Dupe V,
Grondona JM and Chambon P. (1996a). Development,
124, 313 ± 326.
Kastner P, Mark M, Leid M, Gansmuller A, Chin W,
Grondona JM, DeÂcimo D, Krezel W, Dierich A and
Chambon P. (1996b). Genes and Dev., 10, 80 ± 92.
Konieczny SF, Drobes BL, Menke SL and Taparowsky EJ.
(1989). Oncogene, 4, 473 ± 481.
Krezel W, Dupe V, Mark M, Dierich A, Kastner P and
Chambon P. (1996). Proc. Natl. Acad. Sci. USA, 93,
9010 ± 9014.
Kruyt FAE, van der Veer LJ, Mader S, van den Brink CE,
Feijen A, Jonk LJ, Kruijer W and van der Saag PT. (1992).
Dierentiation, 49, 27 ± 37.
Lassar AB, Thayer MJ, Overell RW and Weintraub H.
(1989). Cell, 58, 659 ± 667.
Leblanc BP and Stunnenberg HG. (1995). Genes and Dev., 9,
1811 ± 1816.
Leid M, Kastner P, Lyons R, Nakshatri H, Saunders M,
Zacharewski T, Chen JY, Staub A, Garnier JM, Mader S
and Chambon P. (1992a). Cell, 68, 377 ± 395.
Leid M, Kastner P and Chambon P. (1992b). Trends Biochem
Sci., 17, 427 ± 433.
Levin AA, Sturzenbecker LJ, Kazmer S, Bosakowski T,
Huselton C, Allenby G, Speck J, Kratzeisen C,
Rosenberger M, Lovey A and Grippo JF. (1992).
Nature, 355, 359 ± 361.
Lohnes D, Mark M, Mendelsohn C, Dolle P, Decimo D,
LeMeur M, Dierich A, Gorry P and Chambon P. (1995). J.
Steroid Biochem. Molec. Biol., 53, 475 ± 486.
Mangelsdorf DJ, Borgmeyer U, Heyman RA, Zhou JY, Ong
ES, Oro A, Kakizuka A and Evans RM. (1992). Genes and
Dev., 6, 329 ± 344.
Mangelsdorf DJ and Evans RM. (1995). Cell, 83, 841 ± 850.
Minucci S, Zand DJ, Dey A, Marks MS, Nagata T, Grippo
JF and Ozato K. (1994). Mol. Cell. Biol., 14, 360 ± 372.
Montarras D, Pinset C, Chelly J, Kahn A and Gros F.
(1989). EMBO J., 8, 2203 ± 2207.
Montarras D, Chelly J, Bober E, Armond H, Ott MO, Gros
F and Pinset C. (1991). New Biol., 3, 592 ± 600.
Nagpal S, Saunders M, Kastner P, Durand B, Nakshatri H
and Chambon P. (1992). Cell, 70, 1007 ± 1019.
Nagpal S, Friant S, Nakshatri H and Chambon P. (1993).
EMBO J., 12, 2349 ± 2360.
Nilsen DA, Chou J, Mackrell AJ, Casaban MJ and Steiner
DF. (1983). Proc. Natl. Acad. Sci. USA, 80, 5198 ± 5202.
Perez A, Kastner P, Sethi S, Lutz Y, Reibel C and Chambon
P. (1993). EMBO J., 8, 3171 ± 3182.
Perlmann T and Jansson L. (1995). Genes Dev., 9, 769 ± 782.
Pfahl M, Tzuckerman M, Zhang XK, Lehman JM, Hermann
T, Wills KN and Graupner G. (1990). Methods Enzymol.,
189, 256 ± 270.
Pinset C, Montarras D, Chenevert J, Minty A, Barton P,
Laurent C and Gros F. (1988). Dierentiation, 38, 28 ± 34.
Pratt MAC, Kralova J, McBurney MW. (1990). Mol. Cell.
Biol., 10, 6445 ± 6453.
Robertson KA, Emami B, Mueller L and Collins S. (1992).
Mol. Cell. Biol., 12, 3743 ± 3749.
Rousselot P, Hardas B, Patel A, Guidez F, Gaken J,
Castaigne S, Dejean A, de The H, Degos L, Farzaneh F
and Chomienne C. (1994). Oncogene, 9, 545 ± 551.
Saitou M, Sgai S, Tanaka T, Shimouchi K, Fuchs E,
Narumiya S and Kakizuka A. (1995). Nature, 374, 159 ±
162.
Samuels HH, Stanley F and Casanova J. (1979). Endocrinology, 105, 80 ± 85.
Sive HL and Cheng PF. (1991). Genes and Dev., 105, 1321 ±
1332.
Sorrentino V, Pepperkok R, Davis RL, Ansorge W and
Philipson L. (1990). Nature, 345, 813 ± 815.
Sucov HM, Dyson E, Gumeringer CL, Price J, Chien KR
and Evans RM. (1994). Genes and Dev., 8, 1007 ± 1018.
Sucov HM, Izpisua-Belmonte J-C, Ganan Y and Evans RM.
(1995). Development, 121, 3997 ± 4003.
Tabin CJ. (1991). Cell, 66, 199 ± 217.
Tapscott SJ, Lassar AB, Davis RL and Weintraub H. (1989).
Science, 245, 532 ± 536.
Vincent S, Marty L and Fort P. (1993). Nucleic Acids
Research, 21, 1498.
Wagner M, Thaller C, Jessel T and Eichele G. (1990). Nature,
345, 819 ± 822.
Wagner M, Han B and Jessell TM. (1992). Development, 116,
55 ± 66.
Weintraub H, Davis R, Tapscott S, Thayer M, Krause M,
Benezra R, Blackwell TK, Turner D, Rupp R, Hollenberg
S and Lassar A. (1991). Science, 251, 761 ± 766.
Willy PJ, Umesono K, Ong ES, Evans RM, Heyman RA and
Mangelsdorf DJ. (1995). Genes and Dev., 9, 1033 ± 1045.
Zelent A, Krust A, Petkovich M, Kastner P and Chambon P.
(1989). Nature, 339, 714 ± 717.
Zhang XK, Homan B, Tran PBV, Graupner G and Pfahl
M. (1992). Nature, 355, 441 ± 446.