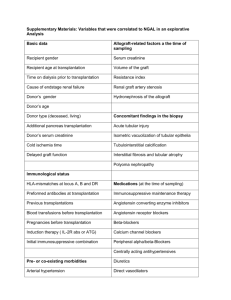
Induction of Immunologic Tolerance for Transplantation
advertisement

PHYSIOLOGICAL REVIEWS Vol. 79, No. 1, January 1999 Printed in U.S.A. Induction of Immunologic Tolerance for Transplantation ALDO A. ROSSINI, DALE L. GREINER, AND JOHN P. MORDES Department of Medicine, University of Massachusetts Medical School, Worcester, Massachusetts I. Introduction A. Overview: from the miraculous to Medawar and Merrill B. The modern era of transplantation C. Beyond immunosuppression II. Transplantation Immunology A. Transplantation terminology B. Immune response to foreign tissues C. Host immune responses to allografts D. Host immune responses to xenografts E. Mechanisms of graft destruction III. Immunologic Tolerance A. New approaches to transplantation B. Definition of tolerance C. Genesis of the concept of transplantation tolerance D. Graft-based tolerance induction E. Host response-based tolerance induction IV. Methods to Induce Transplantation Tolerance In Vivo A. Methods successful in rodents B. Methods successful in large animals and primates C. Evidence of tolerance induction in humans V. Contemporary Clinical Organ Transplantation A. Criteria for successful transplantation program B. Issues of graft placement C. Immunosuppression VI. Transplantation After Tolerance A. Demand for transplantation services B. Potential pitfalls C. Issues of donor tissue and organ availability D. Special case of xenografts E. Conclusion 100 100 100 101 101 101 101 103 106 106 109 109 109 109 110 112 118 118 125 126 126 127 127 127 127 128 128 128 129 129 Rossini, Aldo A., Dale L. Greiner, and John P. Mordes. Induction of Immunologic Tolerance for Transplantation. Physiol. Rev. 79: 99–141, 1999.—In the second half of the 20th century, the transplantation of replacement organs and tissues to cure disease has become a clinical reality. Success has been achieved as a direct result of progress in understanding the cellular and molecular biology of the immune system. This understanding has led to the development of immunosuppressive pharmaceuticals that are part of nearly every transplantation procedure. All such drugs are toxic to some degree, however, and their chronic use, mandatory in transplantation, predisposes the patient to the development of infection and cancer. In addition, many of them may have deleterious long-term effects on the function of grafts. New immunosuppressive agents are constantly under development, but organ transplantation remains a therapy that requires patients to choose between the risks of their primary illness and its treatment on the one hand, and the risks of life-long systemic immunosuppression on the other. Alternatives to immunosuppression include modulation of donor grafts to reduce immunogenicity, removal of passenger leukocytes, transplantation into immunologically privileged sites like the testis or thymus, encapsulation of tissue, and the induction of a state of immunologic tolerance. It is the last of these alternatives that has, perhaps, the most promise and most generic applicability as a future therapy. Recent reports documenting long-term graft survival in the absence of immunosuppression suggest that tolerance-based therapies may soon become a clinical reality. Of particular interest to our laboratory are transplantation strategies that focus on the induction of donor-specific Tcell unresponsiveness. The basic biology, protocols, experimental outcomes, and clinical implications of tolerancebased transplantation are the focus of this review. 0031-9333/99 $15.00 Copyright q 1999 the American Physiological Society / 9j0d$$ja01 P26-8 12-09-98 00:40:31 99 pra APS-Phys Rev 100 ROSSINI, GREINER, AND MORDES I. INTRODUCTION A. Overview: From the Miraculous to Medawar and Merrill 1. Cosmas and Damian rejected a graft from the same donor, or of a first graft in a recipient that had been preimmunized with material from the same donor. 6) The closer the ‘‘blood relationship’’ between donor and recipient, the more likely is graft success. 4. Transplantation genetics and immunosuppression Human history is replete with chimeras, from sphinxes to mermaids, and one wonders if the ancients might actually have dreamed of what we now call xenotransplantation. Certainly they must have dreamed of replacing limbs and other body parts lost to trauma. The first recorded expression of that dream appears in the third century legend of Saints Cosmas and Damian (173, 371). The story is told that these physician brothers used the limb of a Moor who had died to replace the cancerous leg of a church sacristan. Paintings by Fra Angelico and others have immortalized the miraculous legend of the saints’ patient who walked about with one white and one black leg.1 2. The first successful transplant There is remarkable evidence that successful transplantation surgery was actually performed by ancient Hindu vaidya, perhaps 2,000 years ago (236a). The Ayurvedic physicians, among them the author Sushruta, reconstructed noses using pedicle flap grafts from the patient’s own forehead (236a). By 1597, the physician Gaspare Tagliacozzi was performing similar reconstructive rhinoplasty using skin flaps from the patient’s arm (371). There were no reports of success using skin from another person, however (236a, 371). Transplantation progressed little until the 20th century. 3. Laws of transplantation By the beginning of the 20th century, advances in surgery, asepsis, and anesthesia had revivified the ancient dreams of Cosmas and Damian. Animal and some human transplantation experiments began to be performed. These suggested that the serious barriers to successful transplantation would not be technical, but biological. George Schöne summarized these insights in 1912. He defined classic ‘‘laws of transplantation’’ that, in the absence of immune intervention, still apply today (371). These are as follows. 1) Transplantation into a foreign species invariably fails. 2) Transplantation into unrelated members of the same species usually fails. 3) Autografts almost invariably succeed. 4) There is a primary take and then delayed rejection of the first graft into an unrelated member of the same species. 5) There is accelerated rejection of a second graft in a recipient that had previously 1 Fra Angelico (1387–1455): Dream of the Deacon Justinian, Museo di San Marco, Florence, Italy. / 9j0d$$ja01 Volume 79 P26-8 It was decades more before the basis for these empirical laws was found in studies of the small but prolific mouse. Clarence C. Little and George Snell (who later won a Noble prize for this work) had the inspiration to develop and analyze ‘‘congenic’’ strains of mice. Congenic strains are genetically identical except at a single chromosomal locus. By constant breeding and testing for allograft acceptance, Little and Snell pioneered the analysis of what came to be called the major histocompatibility complex (MHC), a genetic locus designated H-2 in mice (371). The MHC is the molecular embodiment of ‘‘blood relationship.’’ Armed with genetic insight, Sir Peter Medawar attempted to apply this information to aid World War II burn victims who could be helped by skin grafts. Like Schöne, he observed that 1) autografts succeed, 2) allografts fail after an initial take, and 3) a second allograft from the same donor undergoes accelerated rejection. He suggested that ‘‘destruction of the foreign epidermis was brought about by a mechanism of active immunization’’ (371). As Medawar continued his analyses using rabbits, his insights led to the recognition that immunosuppression might overcome the laws of transplantation. The development of immunosuppressive agents like nitrogen mustard and corticosteroids and their evaluation in animal models soon led to the practical application of transplantation as a medical therapy. In 1954, Merrill, Murray, and Harrison performed the first successful human vascular organ graft, a kidney transplant (253). The donor and recipient were monozygotic twins in this ‘‘proof-of-principle’’ intervention, with genetic identity obviating the need for immunosuppression. The graft survived until the death of the patient 7 yr later of heart disease. With the advent of an effective immunosuppressive regimen, the same group went on in 1959 to perform the first kidney graft between unrelated individuals; that graft survived for 20 yr (252). B. The Modern Era of Transplantation The modern era of transplantation has also benefited from improved therapeutics for those awaiting transplantation, making them better surgical candidates, improved methods of donor organ preservation, and broadening societal awareness of the need for organ donation. Above all, however, the key to successful transplantation has been the discovery and improvement of strategies for immunosuppression. Radiation of the host came first, 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION but was rapidly supplanted by immunosuppressive drugs including glucocorticoids, azathioprine, and antilymphocyte serum. Newer immunosuppressive drugs with greater potency and wider margins of safety have improved the outcome of renal allografts and generated the first consistent success with cardiac, liver, lung, and pancreas grafts (138). The first of these second-generation drugs was cyclosporine. Newer agents include tacrolimus (formerly called FK-506), monoclonal antibodies like anti-CD3, and many other biologic (393) and nonbiologic agents (262). C. Beyond Immunosuppression The success achieved by solid organ transplant surgeons since the pioneering work of the 1950s has been extraordinary. More than 90% of living related donor and 80–85% of cadaveric kidney grafts are now functional after 1 yr (279, 294). Comparable success has been achieved in the areas of heart, lung, liver, and to a lesser extent pancreas transplantation. This success, however, belies some important residual problems. At this time, all successful treatment of human disease by transplantation (other than between monozygotic siblings) requires the use of general immunosuppressive agents (138). All such drugs are toxic to some degree, and toxicity often leads to patient noncompliance. The drugs are also known to predispose to both infection and neoplasia. Some 10–45% of persons who are chronically immunosuppressed after transplantation develop a neoplasm after 10 yr, and 40–75% do so after 20 yr (75, 272). In addition, many immunosuppressive drugs have deleterious long-term effects on the function of transplanted organs (138), e.g., b-cells in the case of transplantation for diabetes and cyclosporine-induced nephrotoxicity in cardiac and liver transplant recipients (410). New immunosuppressive agents are constantly under development (262, 393), but transplantation of any organ or tissue into an unrelated human currently requires patients to accept the risks of life-long systemic immunosuppression. Another critical problem is that of ‘‘chronic rejection.’’ Although the majority of renal grafts are functional at 1 yr, only 20% will be functional at 10 yr (294). The pathology and mechanisms of this indolent form of graft failure are only beginning to be understood (15, 145, 146, 298). A final issue is recurrence of disease in a successfully transplanted organ. Recurrence may take the form of atherosclerosis in a cardiac allograft or recurrence of tissuespecific disease processes, for example, recurrent autoimmune destruction of pancreatic islets or recurrent lupus nephritis. Finding alternatives to immunosuppression that will prevent both immediate and long-term graft rejection will require further understanding of the mechanisms by / 9j0d$$ja01 P26-8 101 which Schöne’s six laws operate. Immunosuppression is based on interference with the immune system. Understanding immunologic processes better may allow us to redirect the immune response to favor graft acceptance. We next review our current understanding of transplantation immunobiology and newer approaches to transplantation. These include modulation of transplant immunogenicity (sects. IIID1 and IIID2), removal of passenger leukocytes (sect. IVA7), transplantation into immunologically privileged sites like the testis or thymus (sect. IIID3), mechanical encapsulation of tissues (199), and the induction of a state of immunologic tolerance. It is the last of these alternatives that has, perhaps, the most generic promise of revolutionizing tissue transplantation. Induction of transplantation tolerance is the focus of work in our laboratory and the primary focus of this review. II. TRANSPLANTATION IMMUNOLOGY A. Transplantation Terminology Transplauten is a Middle English agricultural word used literally to describe the removal of a plant from one place to another. Its modern surgical adaptation refers to the transfer of an organ or tissue from one part of the body to another or from one individual, the donor, to another, the recipient. If donor and recipient are the same individual, a graft is autologous. If donor and recipient are monozygotic, a graft is syngeneic. If donor and recipient are any other same-species individuals, a graft is allogeneic. If the donor and recipient are of different species, a graft is xenogeneic (174). Orthotopic grafts are placed in the normal anatomic location of that organ; heterotopic grafts are located in other sites of convenience, e.g., intrahepatic placement of pancreatic islets. If a graft heals and functions, it is accepted; if destroyed by the immune system, it is rejected. Rejection may be hyperacute (within minutes to hours) or acute (usually within the first month after grafting). Rejection can also be chronic, with slow, gradual destruction over months to years. If the immune system has not previously been sensitized to the donor’s tissues, the rejection is termed a first-set rejection. In presensitized recipients, the rejection is a second-set rejection. B. Immune Response to Foreign Tissues Many processes participate in the response to all foreign tissue grafts. These include the local inflammatory response to surgery, the processes that initiate wound repair and vascular endothelialization, and the immune response to the recognition of nonself antigen. The basic elements of this response are schematized in Figure 1. 12-09-98 00:40:31 pra APS-Phys Rev 102 ROSSINI, GREINER, AND MORDES Volume 79 FIG. 1. Overview of host immune response to foreign tissue. Host immune response is mediated by mononuclear cells composed of CD4/ and CD8/ T cells (T), macrophages (Mf), natural killer cells (NK), and B cells (B). CD8/ T cells interact with major histocompatibility complex (MHC) class I plus peptide present on graft, including its endothelial cells; CD4/ T cells interact with MHC class II plus peptide present on both graft and endothelial cells. Endothelial cells are depicted as activated in response to inflammatory cytokines, expressing surface molecules that include both MHC molecules and adhesion molecules. Bottom depicts mechanisms of graft destruction associated with various responding host immune cells. Finally, also depicted is possibility of deviation of immune response by Th2-type cytokines produced by CD4/ T cells. ADCC, antibody-dependent cell-mediated cytotoxicity. The response to nonself antigen involves both cellular and humoral immunity. The goal of the response is to reject that antigen, and its nature and intensity are determined by two factors. The first is the biology of the foreign tissue, whether it is an allograft or a xenograft, a vascularized organ or dispersed tissue, a fresh tissue or one that has been pretreated to reduce antigenicity. The second factor is the host response to the encounter with that specific foreign tissue. These responses fall into three categories: hyperacute, acute, and chronic rejection. 1. Hyperacute rejection of discordant xenografts ‘‘Natural antibodies’’ in human recipients cause hyperacute rejection of ‘‘discordant’’ vascularized xenografts (67, 109, 328, 426). These predominantly IgM antibodies recognize a-galactosyl residues that are absent on human (and all old-world primate) endothelial cells but present on endothelium in many other species. They bind to xenogeneic endothelium and fix complement, leading to vascular leakage and thrombosis. The process can cause xenograft rejection in minutes to hours. Preformed anti-a-galactosyl natural antibodies are a major obstacle to the transplantation, for example, of porcine xenografts into humans (128, 149). In contrast, transplantation of allografts never encounters the obstacle of hyperacute rejection due to anti-a-galactosyl natural antibodies. Hyperacute rejection is, however, a well-recognized risk in the case of allografts transplanted into sensitized recipients who have preformed antibodies against allogeneic human leukocyte antigens (HLA). Anti-HLA antibodies occur in individuals exposed to non-self HLA antigens by blood / 9j0d$$ja01 P26-8 transfusion, pregnancy, prior transplantation, or bacterial infections that induce cross-reacting antibodies to HLA epitopes (170). Based on the presence or absence of a-galactosyl residues or anti-a-galactosyl natural antibodies, xenografts can be classified into two groups (11, 343). Concordant xenografts do not elicit hyperacute humoral rejection responses. This occurs most often when donor and recipient are members of closely related species (e.g., rat and mouse). Discordant xenografts do elicit hyperacute rejection responses and commonly occur when donor and recipient are members of less-related species. Strategies for overcoming hyperacute rejection include genetic engineering of xenograft donors to express human complement inhibitors like CD46, CD55, and CD59 (67, 280, 338) or to knock out genes required for the expression of a-galactosyl residues (228, 441). Another approach is prophylactic pretreatment of the human xenograft recipient, for example, by plasmapheresis to remove natural antibodies or by injection of soluble inhibitors of complement such as CD35 (110, 228). Other proposed therapies address events downstream of natural antibody binding and include the administration of antioxidant drugs to maintain the platelet-inhibiting enzyme ecto-ATPase in its active form (67, 280, 338) and depletion of complement by cobra venom. Because none of these promising strategies for overcoming hyperacute rejection involves the induction of tolerance, they are not discussed further. 2. Acute rejection In the absence of a hyperacute rejection response, all transplanted tissues, except for those from identical 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION siblings, engender an acute rejection response. This response is based on the recognition of the foreign tissue as nonself. The basis for recognition of self resides with a system of cell-surface glycoproteins. All living tissues express families of cell-surface proteins that comprise the MHC. For historical reasons, MHC proteins are given the prefix H2 in mice, RT1 in rats, and HLA in people. Alleles of MHC proteins are extremely polymorphic, and chance MHC identity between individuals other than monozygotic siblings is extremely rare. When an allograft is transplanted, the disparity in MHC, or ‘‘alloantigenicity,’’ constitutes recognition of nonself and incites a number of reactions. The interaction of graft alloantigens with the host immune system leads to the activation of several classes of cells. In the first stages of transplantation, as the organ becomes vascularized, there is rapid infiltration of host immune cells into the graft. The infiltrate is composed mostly of mononuclear cells. Although the exact composition of the infiltrate over time is not precisely known, it is presumed that those host mononuclear cells with the capacity to present nonself antigens to the host predominate. Details of this process are presented in section IIC2. 3. Chronic rejection The third major form of graft rejection occurs months to years after transplantation, even in the presence of continued immunosuppression. The majority of kidney transplants, which now have an initial acceptance rate of ú90%, inevitably fail due to the development of chronic rejection and loss of function (279, 294). The pathological hallmark of an organ undergoing chronic rejection is fibrosis, leading to distortion of normal architecture. The pathology is comparable to that which accompanies wound healing, and it has been suggested that chronic rejection is the end result of healing in response to recurring episodes of acute rejection. Other suggested mechanisms include delayed-type hypersensitivity with activation of T-helper cells and B cells (346) leading to activation of macrophages and the secretion of tissue growth factors. Another possibility is antibody-mediated humoral rejection (see sect. IIE10) or endothelial cell damage leading to ischemia (1). In addition, nonimmunologic factors, e.g., ischemia and vascular injury, may contribute to chronic rejection. These factors include maladaptive responses to prior acute injury. The basis of the development of chronic rejection is unknown, and currently available therapies generally fail to reverse chronic rejection, eventually leading to graft failure (15, 146, 298). 4. Graft versus host disease Immune responses to foreign tissue are not totally unidirectional. Essentially all organ and tissue grafts carry with them elements of the host immune system. In the / 9j0d$$ja01 P26-8 103 special case of lymphohemopoietic grafts, the immune system components of the graft are able to mount an immune response to the host. This is termed graft versus host disease (GVHD). In the case of solid organ grafts, however, GVHD is not a typical complication. Donor immune cells, may, however, persist at low levels in the recipient, leading to a state of ‘‘microchimerism.’’ 5. Microchimerism It has been suggested that passenger leukocytes present in at least some successful allografts migrate after organ transplantation and produce persistent chimerism (385). The level of engraftment is low, but it has been argued that a microchimeric state may be essential for sustained survival of allografts (385). The intentional augmentation of microchimerism by cotransplantation of bone marrow together with an organ graft to induce tolerance (46, 276, 446) is discussed in section III. C. Host Immune Responses to Allografts 1. Role of the major histocompatibility complex As noted above, the host MHC plays the defining role in acceptance or rejection of a graft. The more closely the donor graft and the host are matched for MHC, the greater the likelihood of the graft acceptance by the host. The family of MHC cell-surface proteins is subdivided into two major subclasses that play pivotal yet different roles in the determination of self versus nonself by the immune system. These are designated as the MHC class I and the MHC class II antigens. Host T cells can recognize a foreign antigen and generate an immune response, only when foreign antigens are ‘‘presented’’ to its T-cell receptor (TCR) by an MHC class I or class II molecule. In most cases, host MHC class I does not readily present antigens derived from a donor graft, although a mechanism for this type of antigen presentation has been proposed (69, 331, 453). In general, MHC class I molecules present self-antigens derived from intracellular degradation of proteins. These processed antigens are expressed in a cleft present in the extracellular domain of the MHC molecule. Presentation of foreign donor antigens and activation of host-reactive T cells occurs through the MHC class II molecule. Antigens taken up by endocytosis and processed by host antigen presenting cells (APC) are readily presented to the host immune system by the host MHC class II molecule (69, 331, 453). In transplantation, this is primarily the presentation of alloantigen or xenoantigen. In addition to MHC antigens, the immune system can also respond to so-called minor histocompatibility antigens present on grafts. Response to these antigens is indolent, and graft rejection based solely on these antigens is 12-09-98 00:40:31 pra APS-Phys Rev 104 ROSSINI, GREINER, AND MORDES slow (360, 432). An example of a minor histocompatibility antigen is the male Y antigen (360). Peptides noncovalently bound to MHC molecules provide the immune system with antigenic targets. In addition, the presence of peptide bound to MHC molecules lengthens the life span of those MHC molecules on the cell surface of the APC (270). Major histocompatibility complex without peptide has a very short life span and is rapidly lost from the cell surface. The predominant response of the host immune system to foreign tissue is not simply to alloantigen but rather to the allo-MHC plus peptide unit. Direct recognition of allo-MHC without bound peptide can occur, however, and T-cell clones reactive to allo-MHC in the absence of peptide can mediate skin allograft rejection (375). The strategies to induce tolerance described in section IV are directed at altering the acute rejection response. In molecular terms, they are directed at redefining the MHC plus peptide complexes present on the graft as ‘‘self’’ by reeducating the host immune response. Volume 79 and then proliferate. These lymphocytes are found in lymph nodes draining the graft site as well as in peripheral blood and spleen. They are directed against graft antigens presented by either MHC class I or class II molecules on APC. The net effect is activation of the immune system and initiation of immune rejection. Graft destruction can be effected by direct cell-tocell contact between activated effector T cells and the targeted graft resulting in delivery of a cytotoxic molecule. Alternatively, graft rejection may be mediated by indirect mechanisms including lymphokine-induced destruction (see sect. IIE). 2. Antigen presenting cells The primary APC in the immune system are dendritic cells and macrophages. These cells are termed ‘‘professional antigen presenting cells’’ because of their ability to provide accessory cytokines and costimulatory molecules needed for initiation of maximal T- and B-lymphocyte immune reactivity. There are many other types of cells that can present antigen to the immune system, but in general, these do not provide all of the factors necessary for the initiation of an immune rejection response. These ‘‘nonprofessional’’ APC include endothelial cells and resting B lymphocytes. Macrophages are among the first APC to infiltrate a graft. They both initiate and mediate the inflammatory process. Macrophages are able to 1) process and present the antigen to the immune system, 2) release monokines that are both chemoattractant and immunostimulatory, 3) release chemokines that are also chemoattractant and proinflammatory, and 4) scavenge debris from necrotic graft tissue. Another class of professional APC, dendritic cells, can be distinguished from monocytes and macrophages by cytochemical, physical, and morphological characteristics. Dendritic cells reside in the interstitial space of many tissues, process and present antigen, interact with and stimulate T cells, and contribute to allograft rejection. It is now recognized that the ‘‘passenger leukocytes’’ responsible for the immunogenicity of many grafts are probably dendritic cells. Treatments that remove or block dendritic cells have been shown to prolong the survival of transplanted grafts (142) (see sect. IVA7). 4. B lymphocytes The role of antibodies in graft rejection is controversial. T lymphocytes alone are sufficient for rejection of a graft. The humoral response to the graft could, however, participate in this process either by synergizing with T cells or by antagonizing them so as to impede rejection. The humoral immune response to a graft generates antibodies that could synergize with the cellular immune system to enhance graft rejection in several ways. First, graft-reactive antibodies may provide an adherence signal that ‘‘marks’’ a graft and directs macrophages, mononuclear cells, and neutrophils to recognize and attack it. Second, bound antibodies could initiate antibody-dependent cell-mediated cytotoxicity (ADCC), a process in which cells like natural killer cells recognize the Fc portion of bound immunoglobulin and, again, attack the marked graft. Third, antibodies may bind to many target antigens, form immune complexes, and impair the function of the graft. Immune complexes are also recognized by macrophages, again enhancing cellular immunity to the graft. Finally, the antibody could bind complement. This process generates two effects. The bound complement binds macrophages through their C3 receptors. This again enhances macrophage targeting of the marked graft. Another effect is the activation of complement and direct complement-mediated lysis of the targeted graft. Alternatively, antibodies to the graft could act to impair rejection. It has been proposed that antibodies can provide a protective effect termed immunologic enhancement, or simply enhancement (see sect. IIIE3). What determines whether antibodies to grafts facilitate destructive mechanisms or instead act to protect grafts from cellmediated destruction is not known. However, as detailed in section IVA6D, this phenomenon has been demonstrated experimentally in islet transplantation. In those studies, pretreatment of islets with anti-MHC class I antibody extended the survival of human islet xenografts in mice (94). The extent to which enhancing antibodies of host origin may contribute to graft survival is not known. 3. T lymphocytes After presentation of graft antigens by donor or host APC, host lymphocytes become sensitized and activated, 5. Direct and indirect antigen presentation pathways In transplantation, activation of the T-cell component of the immune response by MHC plus peptide can occur / 9j0d$$ja01 P26-8 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION 105 FIG. 2. Direct and indirect pathways of antigen presentation. In the direct pathway, donor peptide is presented to host immune system by donor-origin antigen presenting cells (APC) present in graft. It is thought to be the primary pathway involved in allograft responses. In the indirect pathway, donor peptide that is shed into extracellular space is endocytosed and presented to recipient immune system by host APC. It is thought to be the primary pathway involved in xenograft responses. TCR, T-cell receptor. in two ways. These are termed direct and indirect antigen presentation (225). These processes are summarized in Figure 2. In direct antigen presentation, the MHC molecules on or within the graft present donor antigens directly to host T cells (23, 349). In indirect antigen presentation, antigens ‘‘shed’’ by the graft are thought to be presented to the host immune system by host APC (12, 24, 117, 208, 405). 6. Direct recognition pathway The direct pathway is believed to be of primary importance in the immune response to allografts (42, 207, 349). All allografts contain passenger APC that are transplanted with the tissue. Their removal in most cases is difficult. These donor-origin APC, like most nucleated cells, constitutively express MHC class I molecules that continuously present self-peptides. In addition, they constitutively express coactivation molecules like CD40 and B7–1/2 (142). Alloantigenic peptides presented by donor class I MHC molecules appear to be key targets of T-cell recognition in allorejection (23, 349). T cells of the CD8/ MHC class I-restricted subset directly engage these peptide-MHC class I complexes on APC, receive costimulatory signals, become activated, and undertake to destroy the target (405). This process was first demonstrated in thyroid transplantation (192, 193) and appears to play an important role in islet allograft rejection (68, 95, 190, 372). The case / 9j0d$$ja01 P26-8 of the islet graft is particularly interesting. The passenger donor-origin leukocytes present in grafted islets are APC that express both MHC class I and class II antigens. Islet cells express only class I MHC and no costimulatory molecules on their surface. Removal of passenger leukocytes from the graft removes APC that express MHC class II and costimulatory molecules. This leaves islets with only MHC class I antigen and no costimulatory molecules. This in turn inhibits the host’s ability to respond to foreign alloantigen on the donor islets by the direct presentation pathway, and the graft survives. Several studies support this concept. In some studies, APC were depleted by culturing islet grafts at 247C or with 95% O2 (189). In others, grafts were treated with anti-MHC class II antibody to remove APC. Still others have used a diet low in essential fatty acids to induce emigration of passenger leukocytes from donor islets (209). Each of these methods was reportedly effective in preventing allograft rejection, demonstrating clearly the important role of donor-origin APC present in a graft. Although direct antigen presentation appears to play a key role in acute rejection, it is less likely to be important in chronic rejection (42). 7. Indirect recognition pathways Indirect antigen recognition is based on the theory that donor graft antigen can be shed, subsequently taken up by endocytosis by host-origin APC, processed, and 12-09-98 00:40:31 pra APS-Phys Rev 106 ROSSINI, GREINER, AND MORDES then presented to the host immune system as a self-MHCplus-foreign peptide unit. In the indirect pathway, host T cells recognize graft alloantigens presented by a host APC. This form of antigen presentation by the MHC class II molecule targets class II-restricted host CD4/ T cells, resulting in their activation in response to the graft. Definitive demonstration of indirect antigen presentation and its role in transplantation rejection has been difficult, but convincing evidence has emerged. Indirect allorecognition appears to have a role in chronic allograft rejection (see sect. IIB3), a phenomenon that is mediated in part by the activation of T-helper cells and of alloantibodyproducing B cells (42, 346). The activation of T cells following antigen presentation by the indirect pathway may be less efficient than that of T cells primed by the direct pathway (346). Furthermore, the T-cell repertoire appears to be restricted following indirect antigen presentation, suggesting that a more limited number of antigens may be present by this pathway (224). The classical mechanism of antigen presentation by the indirect pathway involves the uptake of exogenous antigen by the host MHC class II molecules on APC and presentation to CD4/ T cells. It has, however, been suggested that indirect antigen presentation to CD8/ T cells can occur, possibly permitting both CD4/ and CD8/ host T cells to be activated by donor grafts in the absence of direct antigen presentation (25, 181). Indirect antigen presentation is thought to be the major pathway involved in the host response to xenografts, the subject to which we turn next. Volume 79 191) have proposed the concept of ‘‘indirect’’ antigen recognition leading to immune activation and rejection based on the interaction of recipient CD4/ T cells with recipient APC presenting graft xenoantigens. The basis of this preference for indirect recognition in the response to xenografts may stem from the inability of the host CD8/ T-cell population to recognize and interact with the xenogeneic class I MHC molecules present on the graft itself. The generation of cytotoxic T cells and the subsequent release of cytokines and free radicals in the vicinity of grafted tissue are thought then to lead to cytotoxicity or apoptosis (see sect.IIE). Studies of cytokine gene expression are consistent with this view. For example, Morris et al. (261), studying xenogeneic porcine proislets in CBA/H mice, have observed that Th2-like CD4/ T cells appear to be preferentially activated in the course of rejection. E. Mechanisms of Graft Destruction There are several mechanisms available to the immune system of a mammalian host to mediate rejection of a foreign tissue. This redundancy presents additional hurdles that must be overcome in the area of transplantation. Grafts can be destroyed either directly by delivery of a ‘‘lethal hit’’ from cytotoxic cells or indirectly by molecules such as cytokines. Both mechanisms involve a broad diversity of elements within the cellular arm of the immune response (333). 1. Initiation of the effector response D. Host Immune Responses to Xenografts 1. Immune rejection of xenografts The cellular immune response to xenografts is distinct from and less well understood than the cellular immune response to allografts (405). Helper and cytotoxic T-cell responses to xenoantigens are generally slower and less intense than are responses to alloantigens (149). The weaker xenoresponse may result from a relative inability of T cells from one species to interact with APC from another (117, 445). Interactions requiring the adhesion of host immune cells to an organ graft may similarly be weaker across species (149). 2. Direct presentation of xenoantigens Studies of xenograft rejection suggest that MHC class II is the predominant antigenic target of the host immune response and that MHC class II-restricted CD4/ T cells are the mediators of graft destruction (180, 238, 421). It has been suggested, in fact, that xenograft rejection may use a CD4/, cytotoxic T lymphocyte (CTL)-independent pathway (117, 149). Lafferty, Gill, and co-workers (48, 117, / 9j0d$$ja01 P26-8 The cell population that appears to be most important in initiating rejection of allografts is the CD4/ T cell (182). Mice lacking CD4/ cells fail to reject allografts, whereas mice lacking CD8/ retain their ability to reject allografts (182). The ability of CD4-deficient mice to retain grafts may, however, be strain dependent (241). To a first approximation, it can probably be concluded that CD4/ T cells can both initiate and mediate allograft rejection, whereas CD8/ T cells are primarily mediators of graft destruction. The mechanisms available to the T-cell immune system to mediate rejection are described next. 2. Cytotoxic T lymphocytes Cytotoxic T lymphocytes are major mediators of allograft rejection. The CTL are classically CD8/ cells that recognize antigen in the context of MHC class I molecules and, therefore, are important in allograft rejection where the graft itself can present alloantigen to the immune system. In addition, however, subsets of CD4/ T cells can also act as CTL (139). There are at least two major mechanisms by which CTL can deliver a lethal hit through direct cell-cell con- 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION tact. One is by interaction of Fas ligand (FasL, CD95L) on the activated CTL with Fas (CD95) expressed on the target cell (see sect. IIE3). The other is by delivery of cytotoxic molecules termed granzymes (perforin and other molecules like granzyme B; see sect. IIE6). There are several key features of CTL-mediated cytotoxicity. These include 1) antigen specificity, 2) a requirement for cell-cell contact for killing, and 3) the ability of CTL to destroy multiple target cells without injury to themselves. Cytotoxic T lymphocyte-mediated cytotoxicity occurs in several steps. The first is recognition of the target and conjugate formation, whereby the CTL binds to the target cell. This binding event leads to activation signals in the CTL. The activated CTL then delivers a lethal hit mediated either by granzymes or through the Fas pathway. After these steps, the CTL is released from its conjugation with the target cell, and the target then undergoes programmed cell death. Efforts in transplantation to prevent many of these steps have been attempted. The most recent experiments have targeted the Fas-FasL pathway. 3. Fas and Fas ligand Fas and FasL (CD95 and CD95L) are members of the tumor necrosis factor (TNF) family of molecules, and defects in this pathway have been implicated in defective apoptosis and autoimmunity in animals. Mice homozygous for the lpr or gld mutations develop lymphadenopathy and suffer from a systemic lupus erythematosus-like autoimmune disease (268). The lpr defect is due to a mutation in the gene encoding Fas, whereas the gld defect is due to a mutation in the gene encoding FasL (232, 408, 433). Mice with lpr and gld mutations cannot mediate cytotoxic activity through the Fas-FasL pathway, and as a result, they do not reject allografts well. The Fas-FasL pathway has been implicated in clonal selection and control of lymphocyte activation (257, 267, 374) as well as in killing mediated by cytotoxic T cells (339). 4. Immunologically privileged sites It has been known for many years that certain anatomic sites are exceptionally permissive to graft acceptance. Islet allografts, for example, are not rejected even in the absence of immunosuppression when they are placed heterotopically in the testis (20, 371). It is now recognized that several of these ‘‘privileged sites’’ are associated with high levels of FasL expression on cells within the site (127). The interaction between FasL on tissues in the site and Fas on responding host T cells leads to destruction of those responding cells and preservation of the graft. 5. FasL-mediated bystander killing An additional role of cytotoxic cells in graft rejection may stem from their ability to self-regulate. T cells acti- / 9j0d$$ja01 P26-8 107 vated in response to allogeneic stimuli are able to lyse bystander host alloantigen-activated T cells in an MHCrestricted manner (377). This process is not dependent on the perforin pathway but rather on the Fas-FasL pathway of cytotoxicity. Cytotoxic T lymphocyte responses to foreign tissues may therefore cause collateral damage to local host Fas-expressing cells (377). The role of bystander-cell killing in graft destruction or in the generation of collateral damage to host tissues is not yet known. 6. Perforin Perforin is a cytotoxic molecule released into target cells by CTL during conjugate formation. The role of perforin in cytotoxicity may be direct, forming holes in the target cell membrane similar to those produced by the complement cascade. Alternatively, the role of perforin may be indirect, by increasing the porosity of the target cell membrane and thereby enhancing the entry of granzymes. The importance of perforin in transplantation has been highlighted by the observation that mice genetically deficient in perforin are deficient in their ability to lyse allo-specific targets in vitro (162). Cytotoxic CD4/ T cells can also mediate cytotoxicity via the perforin pathway (444). 7. Natural killer cells Natural killer cells (NK cells, sometimes designated as large granular lymphocytes), like CTL, kill target cells using perforin, granzymes, and proteoglycans. The NK cells lyse target cells in an MHC-unrestricted manner using receptors that are not antigen specific. The role of NK cells in allograft rejection is uncertain. The NK cells by themselves cannot reject allografts, but they are in the graft-infiltrating cell population and may contribute to graft damage. The NK cells have been implicated in the rejection of xenografts (219). In the absence of T cells and antigraft antibody, athymic rats reject hamster heart xenografts. Depletion of NK cells by injection of anti-asialo GM-1 antiserum delays rejection, suggesting that NK cell-mediated cytotoxicity can be important in xenograft rejection (219). 8. Macrophages As outlined in section IIC2, macrophages may play multiple roles in initiating and propagating the immune response to grafts. Indirect evidence suggests that they can also mediate graft rejection, at least for xenografts (219). This conclusion is based on several observations. 1) Spleens from animals that are rejecting xenografts harbor a high percentage of macrophages. 2) Rejected xenografts are infiltrated predominantly by macrophages. Furthermore, macrophages adoptively transferred to athymic 12-09-98 00:40:31 pra APS-Phys Rev 108 ROSSINI, GREINER, AND MORDES recipients produce accelerated rejection of newly transplanted xenografts. 9. Dendritic cells Dendritic cells are among the most potent APC and play an important role in both direct and indirect antigen presentation. Dendritic cells, however, are also known to express FasL and can kill activated Fas-expressing CD4/ T cells (400). This activity may contribute to the resistance of a graft to host immune-mediated killing. In addition, dendritic cells can both activate and suppress the host immune system as a function of their expression of FasL or costimulatory molecules (230). The presence or absence of dendritic cells in a graft may be a primary determinant of its acceptance (see sect. IVA7). 10. B lymphocytes As discussed in section IIC4, B lymphocytes and antibody responses have the potential to play many roles in graft rejection. This has become a matter of increasing concern with the recognition that ¢50% of patients on transplantation waiting lists exhibit a high ‘‘panel-reactive antibody’’ status (340). These high levels of antibody to multiple MHC antigens usually result from the blood transfusions patients receive in the course of treatment for their primary disease. They can also arise from pregnancy, prior transplantation, or bacterial infections that induce cross-reacting antibodies to HLA epitopes (170). As noted in section IIB1, this high level of antibody reactivity can lead to hyperacute rejection and therefore poses an additional obstacle to transplantation that must be overcome. High panel-reactive antibody status can be overcome either by selecting the donor graft on the basis of nonreactivity to recipient preformed antibodies or possibly by immunoabsorption of the host’s preformed antibodies before transplantation. Recent studies in animals suggest that presensitization by transfusion can also be prevented. Mice transfused with donor cells in the presence of CTLA4-Ig, a molecule that blocks costimulation, failed to generate antidonor antibodies capable of mediating accelerated rejection (340). To prevent sensitization, current clinical protocols for the care of transplant candidates call for removal of white blood cells before blood transfusion or concurrent treatment with immunosuppressive agents. 11. Natural antibodies and complement Natural antibodies appear to have a role only in xenograft, not allograft rejection. The predominant natural antibodies are those directed against the a-galactosyl residues present on many nonprimate mammalian endothelial cells. They are responsible for hyperacute rejection as discussed in section IIB1. / 9j0d$$ja01 P26-8 Volume 79 Complement appears to have a role in both allo- and xenograft rejection. It can perforate the target cell membrane and create a lethal electrolyte imbalance. Alternatively, it can complex with bound antibody and form a potent adhesion complex for the binding of macrophages and neutrophils, thus targeting these cells to the graft. These functions of complement depend on the generation of an immune response to the graft that leads to antibody formation and antibody-complement complex formation. 12. Cytokines Cytokines can play both destructive and immunomodulatory roles in graft rejection (264). Cytokines that participate in rejection include TNF-a, interferon-g (IFNg), and interleukin (IL)-1 (8, 273). They contribute to graft destruction either directly or by activating effector cells. Cytokines thought to be capable of impairing graft rejection include IL-4, IL-10, and transforming growth factorb (TGF-b). A contemporary controversy in immunology and transplantation concerns the roles of specific T-cell subsets, termed Th1- and Th2-type T cells, in graft rejection (4). The Th1-type CD4/ T cells are producers of IL-2, IFNg, and TNF-b (also known as lymphotoxin). These cytokines can activate both T cells and macrophages. They can promote cellular immune responses that serve as terminal effector mechanisms in allograft rejection. It has also been suggested that expression of the IL-12 receptor b2-subunit is specifically restricted to the Th1-type of cells (332, 406). Interleukin-12 is a proinflammatory cytokine produced predominantly by activated macrophages. The Th1-type immune responses are proinflammatory, promoting CTL development, delayed hypersensitivity responses, and the production of the IgG2a antibody that is involved in ADCC. In contrast, Th2-type cells produce IL-4, IL-5, and IL10, cytokines thought to have immunosuppressive or downregulatory effects on the immune system. It has been suggested, however, that in the absence of CD8/ T cells, CD4/ T-cell production of Th2-type cytokines can also mediate graft rejection (56). Whereas Th1-type immune responses are proinflammatory, Th2-type immune responses are biased toward humoral, IgE-mediated allergic, and mucosal immune responses (264). The Th1- and Th2-type cytokines are not simply the end products of an immune response; they can also, depending on the sequence and intensity of their production, determine (or ‘‘polarize’’) the nature of an immune response. T cells exposed to antigen (e.g., grafts) in the presence of IL-4 are driven toward Th2-type immune responses. In contrast, exposure to antigen in the presence of IFN-g directs T cells toward a Th1-type response. The IFN-g accomplishes this polarization by preventing the differentiation of naive cells to Th2-type cells despite the 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION presence of IL-4; IL-12, when available, then drives the cells toward a Th1-type response (406). Interleukin-12 appears to be a potent determinant of Th1- versus Th2-type polarization of the immune response to foreign tissue. 13. Current status of the Th1-Th2 paradigm The Th1-Th2 paradigm as formulated in mice, however, has begun to break down in its translation to the human immune response. For example, the expression of IL-10 seems to be restricted to Th2 clones in mice, whereas in both humans and rats, IL-10 is expressed by both Th1 and Th2 clones. Moreover, cells other than CD4/ T cells can express IL-10 (79). Mosmann and Kelso suggest that the attribution of graft acceptance versus graft rejection to predominance of a Th1- over a Th2-type immune response may represent an oversimplification of in vivo events (167a, 264). They point to the redundancy of cytokines able to mediate a particular effect and the pleiotropic nature of individual cytokines. The Th1-Th2 paradigm nonetheless remains useful in the interpretation of many aspects of the immune response to foreign tissues. 109 sence of a detectable immune response to a functional graft in the absence of immunosuppression. The functional definition of tolerance is based on graft outcome. Tolerance in immunologic terms, however, may not be induced in all recipients of successful grafts. There are many reports of animals and humans bearing intact and functional grafts in the absence of immunosuppression while at the same time retaining demonstrable host versus graft immune reactivity (119, 385). In addition, a common feature of grafts in ‘‘tolerized’’ recipients is the presence of a mononuclear infiltrate (120, 380). It is unclear whether these infiltrates are destructive or protective, and it is possible that these cells play a role in the maintenance of the tolerant state. The three functional hallmarks of immunologic transplantation tolerance are 1) the lack of demonstrable immune reactivity to graft alloantigens, 2) the presence of immune reactivity to other alloantigens, and 3) absence of generalized immunosuppression for graft maintenance. The immunologically tolerant graft recipient retains a functional graft, retains immune reactivity to all other foreign antigens, and avoids the risks associated with generalized immunosuppression. III. IMMUNOLOGIC TOLERANCE C. Genesis of the Concept of Transplantation Tolerance A. New Approaches to Transplantation The immune response to allo- and xenografts reviewed above is remarkably complex and redundant. Indeed, even drugs that induce a state of general immunosuppression by interfering with many elements of the immune system are still incapable of preventing rejection when MHC mismatches are extensive. It is no wonder that precisely targeted, limited interventions capable of inducing graft acceptance have proven elusive. There remains no question, however, that alternative approaches to transplantation that circumvent the morbidity and mortality inherent in generalized immunosuppression are required. Based on the recent discoveries in immune system recognition and activation, an appealing alternative approach to transplantation is the induction of tolerance to the foreign tissue. Tolerance induction is viewed by many as the most promising alternative to immunosuppression (93, 149, 314, 342, 373, 420). B. Definition of Tolerance Transplantation tolerance may be defined in two complementary ways. It is defined ‘‘clinically’’ or ‘‘functionally’’ as the survival of foreign (allogeneic or xenogeneic) tissue in normal recipients in the absence of immunosuppression. It is defined ‘‘immunologically’’ as the ab- / 9j0d$$ja01 P26-8 One of the first insights into the immunologic basis of transplantation tolerance was the description by Owen (294a) of freemartin cattle. These cattle are not monozygotic, but because of placental anatomy, they share a common circulation during gestation. Owen’s observation was that pairs of freemartin cattle mutually accept allografts without the need for immunosuppression. Billingham et al. (27) then extended this observation by demonstrating that graft tolerance could be induced by exposure of neonatal animals to allogeneic cells. Based on these and other observations, Burnet (47) proposed the clonal selection theory to explain how the immune system distinguished self from nonself. Clonal deletion remains one of the major theories of immunologic tolerance today. Burnet’s theory proposes that clonal selection deletes all self-reactive immune cells in central lymphoid organs like the thymus before their release into the circulation. This theory predicts that all self-reactive cells are deleted before release into the periphery and, by definition, all activation of the normal peripheral immune system must be in response to the detection of nonself antigens. A corollary of the Burnet theory would be that elimination or reeducation of the subset of immune system cells that recognize a specific foreign graft should permit long-term tolerance of a graft in the absence of immunosuppression. It is now recognized, of course, that clonal deletion alone cannot explain all aspects of self-tolerance. Self- 12-09-98 00:40:31 pra APS-Phys Rev 110 ROSSINI, GREINER, AND MORDES reactive lymphocytes and autoantibodies have been detected in circulation of healthy individuals. These observations have required the development of new (but complementary) theories of tolerance based on suppression, lack of appropriate costimulation, and microenvironmental factors that prohibit self-reactive cells from mediating an immune response. These newer theories, in turn, have suggested additional potential mechanisms for inducing long-term tolerance of a graft in the absence of immunosuppression. Most such theories have continued to be based on the Burnet self-nonself paradigm, but the concept of the immune response as recognition of ‘‘nonself’’ has recently been reinterpreted in terms of response to the recognition of ‘‘danger’’ (249). This theory holds that when tissue is damaged (e.g., by infection), molecular signals inform the immune system that danger is present and an appropriate local response is mounted. The theory is based largely on the evolutionary argument that tissue damage would indicate an infection and therefore require an immune response to eliminate the infectious agent. Resolution of the infection would halt tissue damage and ensuing danger signals, and the immune response would recede. This theory predicts that well-healed, quietly functional grafts may not generate danger signals and may be able to survive in the absence of immunosuppression. The richness of the immune response and the theories that have developed to explain self-nonself discrimination suggest that a broad range of strategies may have the potential to induce long-term tolerance of a graft in the absence of immunosuppression. These theories and the strategies that they have inspired are discussed in section IIID. D. Graft-Based Tolerance Induction 1. Fetal tissue and MHC knockouts One obvious strategy for the induction of tolerance to a graft is to reduce its antigenicity, i.e., its ability to provoke a host immune response. Many approaches to the reduction of graft antigenicity have been investigated. The focus of most interventions has been the donor MHC. Much research has centered on the use of fetal tissues because they express lower levels of MHC antigens than do adult tissues (386). Because of the highly experimental nature of the concept and the unresolved ethical issues that surround it, few transplants of fetal human tissues have been attempted. One example is the use of fetal tissues to treat Parkinson’s disease (3, 178, 179). The available animal data suggest that, although the antigenicity of fetal tissues may be less than that of corresponding adult tissues, the reduction in antigenicity is not enough by itself to ensure permanent graft survival (237, 239, 386). The concept remains intriguing, however. In / 9j0d$$ja01 P26-8 Volume 79 animal research, proof-of-concept experiments have used knockout mice that do not express any MHC class I or MHC class II molecules (130), but graft survival was not permanent. Application of this technique to humans would require the development and implementation of xenogeneic knockouts and protocols for transplanting genetically manipulated animal organs. 2. Masking Another approach to modulating the antigenicity of the donor graft has been to ‘‘mask’’ the relevant immunologic recognition sites on transplanted tissues. Human pancreatic islet xenografts have been successfully transplanted into mice without immunosuppression simply by coating the donor islets with F(ab*)2 fragments of antiHLA class I antibody or antibody to tissue-specific epitopes (94). To date, however, no clinical reports of the successful use of this approach in humans have appeared. 3. Privileged sites Immunologically privileged sites in hosts that permit transplantation of grafts without the requirement for immunosuppression have been recognized for more than 50 yr (115, 275, 371). Immunologically privileged sites include the brain, testis, mammary and subcutaneous fat pads, thymus, anterior chamber of the eye, matrix of hair follicles, and the uterus during pregnancy. In Syrian hamsters, the cheek pouch has also been investigated as an immunologically privileged site. Many explanations of immunologic privilege have been offered. These have included a blood-tissue barrier that prevents access by the immune system to the site. This explanation is particularly relevant to privileged sites in the central nervous system. Other explanations have included 1) high concentrations of suppressor cells at the site, 2) soluble molecules secreted by the tissue that modulate local immune responses, and 3) imprisonment of graft antigens within the site, precluding a peripheral immune response. Immunologically privileged sites can also exhibit at least two characteristics that differentiate them from other sites. First, extracellular fluid within these sites generally does not drain directly via lymphatics into lymph nodes, the site where host immune responses are typically initiated. Second, the extracellular fluid in such sites can contain high concentrations of cytokines such as TGF-b, a potent immunomodulatory cytokine that directs immune responses to nondestructive Th2-type rather than Th1type responses. Additional factors must, however, be involved, because infection in the privileged site does induce effector cell infiltration and local immune system activation. In addition, many grafts in privileged sites will survive de- 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION spite the presence of an infection and nonspecific inflammation in the site. At least four additional immunologic mechanisms have been invoked to account for the increasingly complex transplantation biology associated with these sites (392). Depending on the site, these include clonal deletion, clonal anergy, immune deviation, and T-cell suppression, each of which is discussed in a broader context below. None of these mechanisms is mutually exclusive. A) ANTERIOR CHAMBER-ASSOCIATED IMMUNE DEVIATION. Transplantation of human corneas does not require immunosuppression, and for this reason, successful grafts of this tissue actually antedate the first human kidney grafts. These transplants were successful, at least in part, due to the presence of anterior chamber immune privilege (392). This system has been studied extensively, and it has been determined that privilege exists in part because antigens introduced into the eye are captured by distinctive APC that migrate via the blood to the spleen. There they generate a systemic immune response that is biased against the Th1-type CD4/ T cells that mediate delayed hypersensitivity and that help B cells to secrete complement-fixing antibodies. These APC do, however, stimulate CD8/ T cells that function as regulatory cells. The term anterior chamber-associated immune deviation has been coined to describe this immune response. It appears to be mediated by antigen-specific regulatory T cells that secrete TGF-b in an autocrine fashion and suppress effector functions of proinflammatory Th-1type CD4/ T cells. Recent data suggest, however, that FasL (CD95L) expression by host cells is also involved in the maintenance of immunologic privilege at this site (126, 394). It is possible that two or more mechanisms may simultaneously be involved. B) ROLE OF FAS-FASL (CD95-CD95L) IN IMMUNOLOGIC PRIVILEGE. Many activated cells express Fas (CD95), a cellsurface molecule belonging to the TNF family (267). When cells expressing Fas encounter cells expressing the counterreceptor FasL (CD95L), the binding of Fas and FasL leads the cell expressing Fas to undergo programmed cell death by apoptosis. The process is thought to be important generally in the modulation and termination of inflammatory immune responses. Because activated graftreactive T cells express Fas, it was hypothesized that immunologic privilege might involve the expression of FasL within the privileged site. The role of Fas-FasL interaction in immunologic privilege was first documented by the discovery that testis allografts survive indefinitely upon transplantation to allogeneic hosts and that this survival is associated with the expression of FasL (22). Testes from mutant gld mice with defective FasL expression do not survive in allogeneic hosts. Further analyses showed that FasL mRNA is constitutively expressed by testicular Sertoli cells and that Sertoli cells from normal mice, but not gld mutant mice (de- / 9j0d$$ja01 P26-8 111 ficient in FasL), are accepted when transplanted into allogeneic recipients. Expression of FasL in the testis is thought to provide immunologic privilege by inducing apoptotic cell death in Fas-expressing recipient T cells that become activated in response to graft antigens (22). C) ARTIFICIAL IMMUNOLOGIC PRIVILEGE. The ability of Sertoli cells to express high levels of FasL (CD95L) has prompted attempts to use them to provide immunologic protection for other transplanted tissues in a kind of artificial immunologically privileged site. A mixture of Sertoli cells with islets before transplantation into the renal subcapsular space reportedly results in islet allograft survival in the absence of immunosuppression (364). Further evidence consistent with a role for FasL (CD95L) expression on graft survival, and with the concept of artificial immunologic privilege, was provided by Lau et al. (204). These authors expressed FasL on myoblasts, mixed the myoblasts with islets, and observed indefinite islet allograft survival in the absence of immunosuppression in chemically diabetic mice. Graft survival was not prolonged by mixtures of islets and unmodified myoblasts or Fas-expressing myoblasts. Close proximity of FasL-expressing myoblasts and grafted islets was required; islet allografts were rejected if the FasL-expressing myoblasts were placed in the contralateral kidney. The data were interpreted to suggest that the FasL signal provided site- and signal-specific protection of islet allografts. The generation and maintenance of FasL-based artificial immunologic privilege is unlikely to depend solely on the FasL signal, however. For example, the site selected for the generation of artificial immunologic privilege affects the ability of FasL-expressing cells to survive. In one study, transfected hamster kidney cells expressing FasL survived indefinitely in the subrenal space but not subcutaneously in xenogeneic nude mouse recipients. This outcome was hypothesized to result from a FasLmediated inflammatory reaction induced when the graft was placed subcutaneously but not subrenally (450). Expression of FasL is known to be able to generate a severe inflammatory reaction (5, 450). Involvement of factors other than the FasL signal in artificial immunologic privilege has also been suggested by Allison et al. (5), who were unable to confirm the report of prolonged survival of subrenal Fas-L/ testis allografts mentioned above (22). Failure of Fas-L/ testis allografts has also been reported by others (386, 387). Other attempts to exploit graft expression of FasL to achieve transplantation tolerance in the absence of immunosuppression have also failed (64, 163). Transgenic mice were engineered to express FasL under the control of the b-cell-specific rat insulin promoter. In three experiments, expression of FasL on islets not only failed to prevent rejection but actually appeared to accelerate graft destruction. Graft destruction was due to expression of Fas 12-09-98 00:40:31 pra APS-Phys Rev 112 ROSSINI, GREINER, AND MORDES Volume 79 by b-cells in the presence of activated T cells, resulting in b-cell self-destruction (64). Furthermore, in the FasLexpressing transgenic mice, a pancreatic granulocyte infiltration developed, suggesting that FasL expression may have proinflammatory activity when expressed in specific tissues (5). These observations have prompted cautious reevaluation of the use of FasL-expressing cells to induce artificial immunologic privilege for transplantation tolerance induction. E. Host Response-Based Tolerance Induction Induction of transplantation tolerance by altering host responses to the graft fall into two general categories, central and peripheral. We discuss each in detail, but before doing so, we review the mechanisms of T-cell activation that are central to all of the strategies in each category. 1. The two-signal hypothesis How immune responses are initiated has been investigated intensively, and historically, many of the investigations have been tied closely to transplantation biology. Nearly 30 yr ago, Bretscher and Cohn (43) observed that presentation of antigen to the immune system was necessary but not in itself sufficient to initiate an immune response, in that case the production of antibody by B cells. Based on this finding, they proposed the concept of functional deletion or ‘‘paralysis.’’ They introduced a model for B-cell activation that featured a requirement for two signals to achieve activation (43). Bretscher and Cohn (43) proposed that recognition of antigen by a B cell (signal 1) sent a signal that, in the absence of any further intervention, would paralyze the cell. To overcome this paralysis in appropriate circumstances, they proposed that simultaneous delivery of a second signal would induce B-cell responsiveness. They hypothesized that signal 2 came from a T cell. Signal 2 is now termed ‘‘costimulation.’’ The ‘‘two-signal’’ concept was extended to T-cell activation some 5 yr later by Lafferty et al. (192), who proposed that T cells require not only the recognition of antigen in the context of MHC (signal 1) but an additional signal (signal 2) to become fully activated (192). They proposed that signal 2 came from hematopoietic cells, specifically APC. The proposal was based on the finding that survival of allogeneic thyroid grafts was prolonged if the tissue was first cultured in vitro for several days to remove passenger leukocytes, i.e., APC. Their observations were quickly extended to islets, and it was shown again that culture to remove passenger leukocytes prolongs allogeneic islet graft survival (21, 411). The two-signal concept was further developed in in vitro experiments, demonstrating that the delivery of sig- / 9j0d$$ja01 P26-8 FIG. 3. Depicted in schematic form are basic principles of T-cell activation in response to presentation of alloantigen by APC. Process occurs in 3 steps. The 1st step is alloantigen recognition: engagement of TCR by MHC-antigen complex on an APC. Engagement of TCR generates signal 1, which then induces CD154 (CD40L) expression on responding T cell. The 2nd step, induction of CD154, permits engagement of CD40 on APC and is termed coactivation. Engagement of CD40, in turn, upregulates costimulatory molecules B7–1 and B7–2. As a consequence of increased expression of B7–1/2, APC reciprocally triggers T cell via CD28, a process termed signal 2 or costimulation. Activated allospecific T cell now becomes an effector cell capable of mediating allograft rejection. In absence of signal 2 or coactivation via CD40-CD154 that precedes it (step 2), T-cell activation does not occur. Also depicted is CTLA4, a second ligand for B7–1/2 that, unlike CD28, is not constitutively expressed on T cells. Role of CTLA4-B7–1/2 interactions in T-cell activation is controversial (see sect. IIIE1C). nal 1 (antigen) to T cells in the absence of signal 2 (costimulation) shut down IL-2 production, downregulated TCR expression, and induced a state of nonresponsiveness termed anergy (159, 265, 330, 352, 355–357). The process of activating T cells by MHC plus peptide (signal 1) and costimulation (signal 2) is now understood at the molecular level and is known to be more complex than originally thought. To achieve full T-cell activation, three (not two) receptor-ligand interactions must occur. The first interaction is antigen-specific binding of the TCR to peptide-MHC complexes (signal 1). The second is binding of CD154 (CD40 ligand) on T cells to CD40 on APC (coactivation). The third is binding of B7–1/2 on APC to CD28 on T cells (signal 2, costimulation). With certain exceptions not germane to our discussion of tolerance, these three interactions characterize the T-cell response to foreign tissue. These three steps leading to T-cell activation are depicted schematically in Figure 3. The MHC-peptide-TCR interaction (signal 1) was described in Figure 2 and section IIC5. The coactivation and costimulatory interactions are described below. A) CD40-CD154 (CD40L) COACTIVATION. Antigen-specific ligation of the TCR with a specific peptide-MHC complex (signal 1) induces the rapid but transient upregulation of CD154 (also called gp39 or CD40 ligand) on the surface of the T cell. The expression of CD154 on T cells then 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION permits the T cell to activate APC (principally the one that is delivering signal 1). This occurs through the interaction of constitutively expressed CD40 on the APC with the newly expressed CD154 on the T cell (197, 277, 295, 381). CD154 is a glycoprotein expressed predominantly on the surface of activated CD4/ T cells (277, 295), but also on other types of activated cells (66, 103, 195). B) B7 – 1/2-CD28 (CD80/CD86-CD28) COSTIMULATION. The engagement of CD40 with its ligand, CD154, results in the rapid upregulation of the B7–1 and B7–2 costimulatory molecules on the surface of APC (66, 103, 168, 319). The upregulation of B7–1/2 (CD80/CD86) comprises the signal 2 postulated by Lafferty et al. (192) to be required for T-cell activation. This signal is actually transduced to T cells by the receptor-ligand binding of newly expressed B7–1/2 on APC with constitutively expressed CD28 on T cells. Encounter of T cells with antigen in the absence of costimulation by B7–1/2 through CD28 results in their failure to become activated (59, 159, 220, 221, 357, 409, 414). These processes are depicted schematically in Figure 3. C) CTLA4-B7 – 1/2 COSTIMULATION. A second receptor for B7–1/2 on T cells has been discovered and designated cytotoxic T lymphocyte antigen-4 (CTLA4) (44, 221, 357). This CTLA4 is upregulated on T cells 48–72 h after their activation by encounter with antigen and CD40-CD154 coactivation. Expression of CD28, in contrast, is constitutive. The CTLA4 binds to B7–1/2 with 10- to 100-fold greater affinity than does CD28 (165, 357). The role of CTLA4 in regulation of T-cell activation is a subject of considerable interest and controversy. CD28 binding to B7–1/2 is generally believed to deliver signal 2 to T cells and complete the process of T-cell activation. CTLA4 binds to B7–1/2 with higher affinity than does CD28 and competes with CD28 for this ligand. Binding of CTLA4 to B7–1/2 is thought by some to deliver additional costimulation and by others to interfere with costimulation and to deliver a downregulatory signal. Many lines of evidence are consistent with the hypothesis that CTLA4 downregulates activated T cells (165, 184, 413, 417). The most convincing evidence has been provided by CTLA4 knockout mice (415, 434). These animals develop a severe lymphoproliferative disease and die early in life. The disease can be prevented by administration of CTLA4-Ig fusion protein, a hybrid molecule that blocks B7–1/2 interaction with CD28. Cessation of CTLA4-Ig treatment leads to recrudescence of the lymphoproliferative disease and death (416). Another study has shown that anti-CTLA4 antibody treatment of young transgenic mice with a diabetogenic TCR is permissive to the expression of autoimmune diabetes (231). Treatment of mice with anti-CTLA4 monoclonal antibody (MAb) has also implicated CTLA4 as an important downmodulator of T-cell activation. Treatment with a blocking anti-CTLA4 MAb is permissive to in- / 9j0d$$ja01 P26-8 113 creased T-cell proliferation (222); a different anti-CTLA4 MAb can induce apoptosis of T cells, the ultimate form of downregulation (125). Ligation of CTLA4 may also deliver a signal that is necessary for tolerance induction, another manifestation of the downregulation of T-cell activation (187, 243, 305). Although these data strongly suggest that CTLA4 acts primarily to downregulate immune responses, other data suggest that the opposite effect may occur in certain circumstances. For example, CD28-deficient mice can activate T cells and induce their proliferation through the CTLA4 molecule (448). Moreover, the antibodies used to define an immunomodulatory role for CTLA4 may in fact be blocking antibodies that prevent the enhancement of T-cell responsiveness by CTLA4 (223). D) THE B7 FAMILY OF COSTIMULATORY MOLECULES. The expression of B7–1 (CD80) and B7–2 (CD86) follow different kinetics. The molecules appear at different times during the development of an immune response (144, 449). Costimulation provided through these molecules may generate different cytokine profiles in stimulated T cells (107, 185). Ligation of CD28 by CD86 appears preferentially to induce expression of IL-4, a Th2-type cytokine, whereas ligation of CD28 by CD80 appears to induce a less biased cytokine profile. The CD80/CD86 system is growing in complexity. Expression of CD80 and CD86 may be differentially regulated by cytokines in the microenvironment in which they interact with CD28 (202). Antibodies to B7–1 and B7–2 have differential effects on immune responses, especially on autoimmune responses (185, 213). In addition, at least three forms of CD80 have been identified in mice (16, 40, 106, 133, 143, 154, 155). Molecules in this B7 family are designated B7–1, B7–1a, and B7–1cytII. These molecules exhibit differential and variable expression, diverse immune response effects, and a range of binding affinities to CD28 and CTLA4. The differential activity of each in relation to the provision of costimulation is yet to be determined (34, 300). For the purposes of this review on induction of transplantation tolerance, the key observation is that blockade of B7-CD28 interaction has potent immunosuppressive and perhaps tolerance-inducing properties in allograft and xenograft transplantation (see sect. IVA8). E) ACTIVATION OF NAIVE VERSUS MEMORY CELLS. We conclude this discussion of the two-signal model with the observation that costimulation (and cytokine) requirements for activation of naive versus memory T cells are very different. The former have never been activated; the latter have been activated by antigen exposure and retain the memory of that exposure. Much more stringent criteria must be met for the activation of naive T cells than for the reactivation of memory cells (41, 87, 403). In addition, as noted in section IIE12, the immune response of T cells following activation can be biased 12-09-98 00:40:31 pra APS-Phys Rev 114 ROSSINI, GREINER, AND MORDES toward either Th1- or Th2-type effectors as a function of cytokine milieu in which they first encounter antigen (402). Together, these observations have practical implications for transplantation tolerance protocols that involve polarization of the immune response toward nondestructive Th2-type reactivity. Naive T cells display higher requirement for antigen, need stronger costimulatory signals, and rely more heavily on the presence of enhancing cytokines for activation than do memory T cells (70). With current technology, it is difficult, if not impossible, to tolerize memory cells in transplantation. As described in section IV, almost all successful tolerance-induction methodologies employ techniques to prevent the activation of naive T cells. If presensitization of the T-cell immune system has occurred due to previous transplants or exposure to multiple transfusions, adjunct therapy to eliminate the preexisting memory T cells is routinely required for tolerance induction. 2. Central tolerance Tolerance to self at the T-cell level occurs during differentiation of T cells in the thymus. In this organ, T cells develop their TCR repertoire, and selection against self-antigen responsive cells occurs by both positive and negative processes. T cells must first recognize self-MHC to permit their survival (positive selection). If the affinity of TCR binding to self-MHC is too strong, however, the cells are thought to be clonally deleted (negative selection). A) CLONAL DELETION. The classical form of endogenous self-tolerance requires the deletion of self-reactive T lymphocytes in the thymus (32, 164). The clonal deletion of developing T cells involves deletion of immature CD4/CD8/ thymocytes after they first express their TCR on the cell surface. This process has been elegantly demonstrated by following the intrathymic development of specific populations of developing T cells that express unique naturally occurring TCR Vb sequences or a transgenic TCR that is directed against self-antigen (172). The clonal deletion mechanism of tolerance postulates that all self-reactive T cells are eliminated before their migration to peripheral tissues. This mechanism predicts that the introduction of transplantation antigen into the thymus should result in the selective deletion of all T cells reactive to that antigen and should therefore induce tolerance. This hypothesis has been tested and, as described in section IVA4, is a potent method for inducing transplantation tolerance. It is unlikely, however, that the immune system relies solely on clonal deletion to maintain tolerance. Self-reactive T cells do, in fact, escape from the thymus to the periphery, but in most individuals, they do not lead to immune reactivity against self. Furthermore, central toler- / 9j0d$$ja01 P26-8 Volume 79 ance cannot explain 1) how self-reactive cells are eliminated if the peripheral antigen is not expressed intrathymically, 2) how tolerance to cryptic intracellular antigens is achieved, or 3) how tolerance is generated to antigens to which the immune system is exposed late in life. Given that that prevention of self-reactivity without compromising the ability to mount a danger response is so critical, it is not surprising that tolerance induction is a redundant process both in the thymus and in the periphery. Immunologists have recently exploited this redundancy to develop multiple approaches to the induction of transplantation tolerance. One such redundant intrathymic mechanism is clonal inactivation. B) CLONAL INACTIVATION. Clonal inactivation is a nondeletional mechanism of self-tolerance that occurs intrathymically (318). Tolerance by inactivation involves radiation-resistant thymic stromal elements, perhaps thymic epithelial cells (150, 353). Self-reactive cells tolerized by this process are described as ‘‘clonally anergic.’’ Anergized, clonally inactivated antigen-reactive T cells persist in the host, but they do not become activated in response to antigen presentation. This form of T-cell nonresponsiveness has been exploited for the induction of transplantation tolerance in the peripheral immune system to which we turn next. 3. Peripheral tolerance Peripheral tolerance is based on the observation that self-reactive T cells that escape into peripheral tissues appear to be held in check by immunomodulatory controls. Multiple components of the peripheral tolerance process have been identified. These have yielded several approaches to the induction of transplantation tolerance. The specificity and safety of each approach have improved steadily. A) ANERGY. As noted above in section IIIE2B, anergy is defined as a state in which T cells are refractory to activation. In the periphery, anergy is thought to occur when T cells bind ligand in the absence of costimulation. Anergic T cells have been documented by demonstrating the presence of cells with a TCR directed toward the antigen of interest that nonetheless do not respond to that antigen in the presence of costimulatory signals. Anergy was first suggested in 1983 (196) and formally demonstrated in 1987 (159, 265). In those studies, antigen was presented to T cells using fixed APC which, because of the fixation, could not upregulate B7 molecules and would therefore not provide costimulation. The defining in vitro characteristic of anergic cells was their inactivation by antigen rather than their deletion. In addition, they lacked IL-2 production (235) and evidenced downregulation of TCR expression (177, 330, 358, 454). Finally, it was also shown that the responsiveness of anergic cells to antigen could be restored (‘‘rescued’’) by incubation in the presence of IL-2 (6, 26, 147). 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION It is thought that anergy can be induced both intrathymically (317, 318) and in the peripheral lymphoid tissues (183, 198, 315, 330). Anergy appears to be much easier to induce in cloned T cells than in fresh antigen-stimulated cells. However, to date, there is no evidence that tolerance by anergy induction can be applied to antigencommitted memory T cells in vivo (436). It has also been suggested that anergic T cells not only fail to respond to antigen upon restimulation but may also function as suppressor T cells (105, 226). The mechanism of this suppression is hypothesized to involve competition for antigen presented on the surface of the APC and competition for locally produced IL-2. As long as antigen-reactive cells are blocked from proliferation, they can act as suppressor cells (359). Peripheral cell suppression and its regulation of immune responses are discussed in section IIIE3C. B) IMMUNOLOGIC EXHAUSTION. Exhaustion is a mechanism of tolerance very similar to anergy, but different with respect to the ultimate fate of the antigen-specific T cell (329). Immunologic exhaustion can be detected in populations of antigen-specific T cells that are initially activated and expanding. Those populations of T cells undergoing expansion while exposed to high concentrations of antigen were found to be inactivated (anergized), whereas T cells exposed to lower concentrations of antigen were observed to disappear (exhaustion). The disappearance of antigen-reactive T cells in this setting may involve apoptosis (30, 140, 217). C) PERIPHERAL CELL SUPPRESSION. There has been considerable controversy over the existence, identity, and function of peripheral suppressor cells. It is not clear if suppression is mediated by a distinct population of cells or is mediated by cells that, under different conditions, can mediate suppression, help, or cytotoxicity. It has even been suggested that suppressor cells can mediate regulation of target cell killing either by cytokine-mediated modulation or cytotoxicity directed against other immunocytes. Two classic examples of possible suppressor cell activity in vivo are based on studies of neonatal tolerance maintenance and of the regulation of autoimmunity. In classic neonatal tolerance induction protocols, neonatal animals are injected with allogeneic donor bone marrow. Not only is the donor bone marrow graft then tolerated, but it can also be shown that tolerance to the donor cannot be broken by infusion of additional nontolerized syngeneic lymphocytes (334). This finding suggested the existence of an active suppressive mechanism because the infused lymphocytes were functionally capable of mediating reactivity against the donor graft. In studies of the regulation of autoimmunity, it has been shown that autoimmune insulin-dependent diabetes mellitus in both rat (259, 260, 335) and mouse (321) models can be modulated by regulatory or suppressor cells. The suppressor hypothesis has been put forward to / 9j0d$$ja01 P26-8 115 explain two important observations in transplantation. The first is that alloreactive cells are present in animals bearing intact grafts (9, 74, 258). In vitro proliferation and cytotoxic activity to the specific tissue alloantigen of the graft can be readily detected in animals bearing intact, functional grafts. The second observation is the presence of cellular infiltrates in grafts that are not rejected and remain functional (9, 334). The suppressor cell hypothesis has also been invoked to explain the apparent induction of in vivo allotolerance by the transfer of suppressor cells to naive animals (infectious tolerance, see sect. IIIE3D). It has also been invoked to explain the inhibition of alloresponsiveness in naive cells that are cultured in vitro in the presence of putative suppressor cells (78, 85, 137, 314, 334, 392). Both CD4/ and CD8/ T cells have been implicated as mediators of peripheral suppression (97, 137, 289, 290). In cases of immunoregulation of autoimmunity that have been studied, the CD4/ cell subset appears most likely involved. In cases of the regulation of cytotoxic T cells, the CD8/ T-cell subset has been postulated to mediate the activity. Several hypotheses have been put forward to explain the mechanism by which suppressor cells regulate immune responses. One hypothesis holds that secretion by suppressor cells of cytokines like IL-4 (312) and TGF-b (327) modulates immune reactivity in the local microenvironment. The possibility that TGF-b functions in this way has been termed ‘‘cytokine-mediated bystander suppression’’ (62, 63, 100, 226, 457, 458). Another possibility is that activated T cells expressing FasL kill all activated T cells at the site of immune reactivity, effectively downregulating the immune response (267, 459). A third alternative hypothesis is that CD4/ suppressor cells mediate their activity by blocking the indirect pathway of alloantigen recognition, effectively preventing priming of the immune system (452). Which of these proposed mechanisms, or which combination of these mechanisms actually mediates suppression is unclear. It is also unclear whether suppressor cells represent a unique population of cells or normal helper and effector cell populations that are exhibiting alternative functionality. Despite these uncertainties, however, the available data strongly support the concept of suppressor cell activity and its importance as a mechanism of tolerance induction. D) INFECTIOUS TOLERANCE. The case for the existence of suppressor cells has received additional support from the demonstration of infectious tolerance. This type of tolerance is characterized functionally by the ability of adoptively transferred CD4/ cells to inhibit the ability of naive lymphocytes to reject grafts (314). Many examples of this type of tolerance have been documented. Most examples invoke infectious tolerance as the mechanism by which anti-CD4 MAb therapy may induce transplantation tolerance (292, 362) (see sect. IVA6A). 12-09-98 00:40:31 pra APS-Phys Rev 116 ROSSINI, GREINER, AND MORDES E) CLONAL DELETION. Although central intrathymic clonal deletion has long been recognized as a primary mechanism of tolerance induction to self-antigens, clonal deletion of peripheral T cells and tolerance induction has only recently been appreciated (435). This mechanism has been elegantly demonstrated in mice using minor lymphocyte-stimulating (Mls) superantigens that are now known to be exogenous mouse mammary tumor viruses (2, 65, 429, 447). In these studies, exposure to Mls antigen led first to an expansion of Mls-reactive cells that was followed by clonal deletion of the Vb6/ cells that recognized the antigen. This mechanism was also functional for CD4/ antigen-specific T cells, demonstrating that the peripheral clonal deletion of Mls-reactive cells is not a unique property of the Vb6/ Mls-superantigen-reactive cell population (198). Clonal deletion of CD8/ cells has also been demonstrated, and cytokines may play a role in this process (361). Specifically, the Th2-type cytokines IL-4 and IL-10 have been implicated in the clonal deletion of alloreactive CD8/ T cells that mediate graft rejection; deletion of reactive cells was confirmed using a clonotypic antibody against the TCR expressed on the transgenic T cells used in the experiment (361). Finally, as discussed in section IIID3B, the interaction between activated T cells expressing FasL (CD95L) and activated cells, including T cells expressing Fas (CD95), has been strongly implicated in the clonal deletion of immune response cells. Deletion of the antigen-reactive cell population is not always required, however, for the induction of a nonresponsive immune state. An example of nondestructive, noninactivating tolerance induction is immune deviation. F) IMMUNE DEVIATION. As was the case for the theory of suppressor cell function in transplantation tolerance, there is controversy regarding the role of immune deviation in allograft tolerance and survival. The controversy stems in part from the large number of factors that can ‘‘deviate’’ the immune response and the diversity of mechanisms that lead to the expression of those factors. The strongest evidence for immune deviation is in the area of control of the self-reactive autoimmune response. In autoimmunity, the deviation of an immune response from the destructive cytokines IL-2, IFN-g, and IL12 to the ‘‘suppressive’’ cytokines IL-4, IL-10, and TGF-b is associated with disease prevention (4). As noted above, this type of deviated cytokine response has also been implicated in neonatal tolerance (60, 102, 111, 334), but the importance of immune deviation in the modulation of graft immune responses is uncertain. One of the best-characterized instances of graft tolerance that may be mediated, at least in part, by immune deviation is the immune privilege of the eye (see sect. IIID3B). In the anterior chamber, it is hypothesized that, following encounter with antigen, T cells become anergic, / 9j0d$$ja01 P26-8 Volume 79 undergo apoptosis, secrete suppressive TGF-b, and release soluble regulatory factors. In addition, APC indigenous to the eye take up antigen and subsequently migrate to the spleen where they activate a unique spectrum of antigen-specific T and B cells. These cells generate a Th2type immune response associated with the absence of delayed-type hypersensitivity and complement-fixing antibodies (392). Immune deviation, however, is only one of at least four possible mechanisms that mediate ocular tolerance, with the others being clonal deletion, clonal anergy, and T-cell suppression (391, 392). Transforming growth factor-b also has been reported to have potent immunosuppressive effects in allograft survival experiments (316, 366, 430). The Th2-type cytokine IL-10 may also play a role in immune deviation-related graft tolerance. Interleukin-10 has many effects that may deviate the immune response. It has been shown to prevent APC activation and costimulation, permitting antigen presentation in the absence of costimulation and adhesion molecule expression (57, 129, 442). It may also induce an anergic state in CD4/ T cells (83), suppress production of the proinflammatory cytokine IL-2 by CD4/ T cells (81), inhibit T-cell proliferation (407), and regulate expression of IFN-g and IL-4 by monocytes, dendritic cells, and Langerhans cells (APC). The role of agents like IL-10-Fc fusion proteins to serve as tolerogenic pharmaceuticals is under study (461). The action of suppressor cells may also involve immune deviation principles. In studies of the adoptive transfer of transplantation tolerance with suppressor cell clones, it has been shown that the clones are Th2-type cells (236). Immune deviation may also occur when costimulation is blocked. Blockade of the CD28-B7 costimulatory pathway after alloantigenic stimulation in vivo inhibits the production of Th1-type, but not Th2-type cytokines (345). In relation to the indirect evidence suggesting that cytokines may downregulate the immune response, a note of caution must be sounded. As Nickerson et al. (273) have recently pointed out, ‘‘It is notable that in no circumstances to date has allograft tolerance been achieved via specifically blocking the production/effects of a proinflammatory cytokine (e.g., IL-2), and/or enhancing the endogenous production or local/systemic administation of a cytokine with immunoregulatory properties (e.g., IL-4, IL10, TGF-b).’’ Whether the cytokines detected at the site of graft acceptance or rejection are causally related to the ongoing local immune response is uncertain. The cytokine profiles could 1) arise as a result of a tolerant state induced by a different mechanism, 2) contribute ancillary support for the maintenance of a tolerant state, or 3) arise independently as a consequence of the tolerant state (308). Many studies argue strongly against a primary role for immune deviation in graft survival. In many cases of tolerance induction to grafts, the cytokine profile exhibits 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION neither a Th1- nor Th2-type bias (161, 201). Many attempts to skew the cytokine immune response and induce tolerance have failed (56, 423, 428, 461). Administration or expression of IL-10 and IL-4 have not been associated with tolerance induction (274, 461). In addition, inhibition of Th1-type cytokines and induction of Th2-type cytokines by blocking IL-12 fail to induce tolerance (307). Interleukin-2, a Th1-type cytokine, has a potentially important role in graft rejection because activated T cells require it for proliferation. Interleukin-2 does, in fact, appear to exert a detrimental effect on transplantation tolerance induction. Administration of IL-2 as an adjunct to various tolerizing regimens appears to preclude long-term graft survival (73). Conversely, blockade of IL-2 binding to its receptor on activated T cells may be a promising approach for induction of transplantation tolerance. AntiIL-2 receptor antibodies are in preliminary clinical trial (273), but a cautious approach is necessary. The theoretical possibility exists that the antibodies will destroy all activated T cells and render the patient susceptible to infection, similar to generalized immunosuppression. In addition, the potential of IL-2 blockade to prevent allograft rejection is uncertain given that IL-2 knockout mice reject islet allografts with the same rapid kinetics as do normal mice (388). These variable and sometimes contradictory observations may be explained by 1) the redundancy in the family of cytokines, with various members acting as T-cell growth factors, and 2) the pleiotropic nature of individual cytokines (273). These biological characteristics have dampened initial enthusiasm for cytokine manipulation and immune deviation as viable approaches to the induction of clinical transplantation tolerance. G) CHIMERISM. As noted earlier, the first reported case of tolerance was in freemartin cattle by Owen (294a). This form of tolerance is based on lymphohemopoietic chimerism. This historic observation has been revisited and exploited to induce transplantation tolerance experimentally in both rodents and primates (see sect. IVB). The mechanism of tolerance of this type requires chimerism in the antigen-presenting cell compartment and, specifically, in the dendritic cell compartment (384, 385). Passenger leukocytes present in a graft are now known to induce a state of microchimerism in the recipient. In these microchimeric graft recipients, donor-tissuespecific hyporesponsiveness has been documented; it appears to correlate with engraftment of donor cells within the transplanted tissue (322, 455) and with graft survival (323). Donor-derived dendritic cells are hypothesized to emigrate from the donor graft and to migrate to lymphoid tissues of the host where the tolerance-inducing effect is mediated (306). Donor bone marrow transplantation has been used as an adjunctive therapy to augment donor-cell chimerism in recipients of kidney, liver, heart, and pancreas trans- / 9j0d$$ja01 P26-8 117 plants (46, 101, 216, 384). More recently, this procedure has been applied to human islet and lung transplantation (53, 306), and its use has been proposed for induction of xenograft tolerance (153). The use of microchimerism to induce tolerance is not without controversy. The available data have alternative interpretations. The controversy revolves around the question of whether the chimerism observed in long-term graft survival recipients is the cause or the consequence graft survival (446). In addition, some investigators have failed to confirm the presence of the peripheral chimerism in long-term graft recipients reported in earlier studies (397). These investigators have offered an alternative explanation for the persistence of donor-origin cells in tolerized graft recipients. They argue that the tolerance observed after donor bone marrow administration is in fact ‘‘high dose, activation-associated tolerance,’’ a form of clonal deletion (29, 263, 435). The postulate in these studies is that graft reactive cells are strongly activated, and this strong activation leads to clonal deletion. Additional experimentation will be required to establish the relative importance of chimerism in transplantation tolerance. H) IGNORANCE. As discussed in section IIIE2A, clonal deletion in the thymus is not a completely effective process. To deal with self-reactive (and by extension graftreactive) T cells, the immune system has developed multiple redundant mechanisms for establishing and maintaining peripheral tolerance (10, 254). ‘‘Ignorance’’ is another such mechanism. The persistence of graft-reactive T cells in recipients of functioning allografts has led to the proposal that the T-cell immune system simply ‘‘ignores’’ the graft. The difference between ignorance and tolerance is the ease with which the nonresponsive state can be broken and the graft rejected. Classical approaches to the breaking of immunologic ignorance include infection (284, 285) and the presentation of graft antigen in combination with professional APC (68). An example of ignorance is provided by transgenic mice with a TCR that recognizes the glycoprotein of lymphocytic choriomeningitis viral (LCMV) and transgenic expression of that glycoprotein in the host’s pancreatic islets by placing the transgene under control of the insulin promoter (285). The antigen-specific T cells in the host fail to mount an immune response against the LCMV protein present on the islets. The initial nonresponsiveness, or ignorance, is thought, perhaps, to be due to the inability of the islets to both present antigen and provide costimulation. However, infection with LCMV leads to a rapid immune response against the live virus and to the destruction of the islet cells expressing the LCMV protein (284). The persistence of the antigen-reactive T cells in a nonresponsive state until active viral infection may mimic the situation in transplantation, where a graft survives in the presence of circulating alloreactive T cells, i.e., the presence of ignorance. 12-09-98 00:40:31 pra APS-Phys Rev 118 ROSSINI, GREINER, AND MORDES I) VETO CELLS. Transplantation tolerance may also involve ‘‘veto’’ cells. Such cells are defined functionally by their ability to serve as targets of alloantigen-specific cytotoxic T cells and then kill the cytotoxic T cell, thus vetoing their own cytolysis (255, 405). The veto phenomenon has been documented in many experimental models, but the mechanism of veto cell modulation of CTL activity is uncertain. Veto activity may be due to the killing of activated CTL, possibly via the apoptosis-inducting function of FasFasL interaction. Cytokines have also been implicated in veto activity. In one primate study, TGF-b has been implicated in the veto of CTL activity by rhesus monkey bone marrow cells (255). J) ENHANCEMENT. Enhancement is antibody-mediated prolongation of graft survival. Experimentally, enhancing antibodies generated against the donor graft have been shown to be effective in prolonging the survival of allogeneic heart grafts in the rat (299) and kidney grafts in the primate (412). Anti-HLA antibodies have been shown to enhance allograft survival (398). Enhancing antibodies appear most effective in the context of islet grafts and least effective in the context of skin grafts (269). At this time, the utility of enhancing antibodies in clinical transplantation is uncertain, and the possibility of antibodies detrimental to grafts has not been excluded. IV. METHODS TO INDUCE TRANSPLANTATION TOLERANCE IN VIVO Volume 79 A. Methods Successful in Rodents As was made clear in sections ID and IE, the two major foci for tolerance-inducing regimens are donor tissue or the host immune system. Each has been manipulated with varying degrees of success, and it is possible that combination methodologies will ultimately be required for successful translation to the clinic. 1. Graft plus generalized immunosuppression Generalized immunosuppression is the universal method for preventing acute graft rejection today. Generalized immunosuppression is also universally prescribed for the maintenance of successful allografts. In theory, however, the microchimerism induced by organ transplantation should permit withdrawal of that therapy. Only a few such instances have been reported. The few cases that have been reported are anecdotal and resulted from noncompliance rather than scientific intervention (385). There is little enthusiasm for a clinical trial of withdrawal of immunosuppression. The consensus opinion is that tolerance is not routinely induced in graft recipients treated with generalized immunosuppression and that continuous therapy is required for graft survival. As detailed above, most tolerance-induction methods require activation, or at least ongoing involvement, of the immune system. Almost by definition, then, the use of generalized immunosuppression to prevent graft rejection will pari passu preclude the induction of tolerance. The dilemma that may soon face clinicians is whether the tolerance-inducing protocols that do not also use generalized immunosuppression are worth the risk. The resolution of that dilemma will be based on the outcome of animal studies, and to those we turn next. The preceding discussion on the theories of graftand host-based transplantation tolerance induction has illustrated the complexity of the subject and the uncertainties faced by clinicians seeking to move transplantation ‘‘beyond immunosuppression.’’ Not surprisingly, the myriad theories and mechanisms invoked to explain toler2. Donor-specific transfusion ance induction and tolerance maintenance have led to myriad approaches to the goal of enhancing the duration Transfusion had its origins in the correction of aneof graft survival by methods that are safe. mia, but it is now recognized that transfusion also affects As we have noted earlier, the most promising alterna- the immune system. Beneficial effects of transfusion on tives to immunosuppression are strategies that seek to the recurrence of cancer, postoperative infection, viral induce donor-specific immune tolerance (131, 218, 314, infection, inflammatory disease, and pregnancy have been 369). Many of those strategies have been discussed above noted (35). As early as 1974, it was recognized that blood in the context of the underlying theory and available evi- transfusion before kidney transplantation improved graft dence. Their application as actual protocols to experimen- survival (293). In that seminal report, it was concluded tal induction of tolerance has been advancing rapidly. that ‘‘transfusions in prospective recipients lead to an imEach of the protocols discussed below has been de- provement in transplant survival, probably because of acveloped explicitly to establish a tolerant state adequate tive induction of immune unresponsiveness.’’ to support the survival of allografts. At this writing, no The use of transfusion in transplantation focuses on tolerance-inducing protocol has been substituted for gen- donor-specific transfusion (DST). Donor-specific transfueralized immunosuppression in the clinic. We first discuss sion has been observed to abrogate the development of the protocols that have been reported to be successful in chronic graft rejection in mouse and rat models of transsmall-animal models and then consider protocols that plantation (80, 151, 248, 266, 294, 379, 396). The mechanism have been successfully adapted to larger animals and underlying the unresponsive state that is induced by DST shown to be effective in primates. has received considerable experimental attention since the / 9j0d$$ja01 P26-8 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION first reports (36, 37). Proposed mechanisms have included immune deviation (37) and the activity of veto cells (86, 167). The improved outcome of graft survival has also been correlated with evidence of donor-specific hyporeactivity (323). Donor-specific transfusion has also been used in combination with ultraviolet irradiation, another inducer of hyporeactivity, to prolong islet allograft survival (203). The tolerance-inducing component of the DST has not been identified with certainty and may be different depending on the extent of donor-host MHC mismatch. In one study, irradiation-sensitive donor T cells (veto cells) were required for tolerance induction to MHC class I antigens, whereas irradiation-resistant non-T cells were required in the DST for tolerance induction to MHC class II antigens (426). One cell type present in DST that has characteristics potentially important for tolerance induction is the small B lymphocyte. Small resting B cells are good candidates for tolerizing T cells because they can present antigen in the absence of costimulation. Small B lymphocytes express only low levels of the B7–1/2 costimulatory molecules on their surface; unless stimulated, they are ineffective as APC (116, 158, 296). Small B cells induce tolerance when acting as APC for protein antigens in vivo. They mediate their tolerizing effects by inducing unresponsiveness in the helper T-cell compartment (91, 92, 98). Small B lymphocytes also induce specific transplantation tolerance to an MHC class II difference (151) and, in unprimed animals, to H-Y, a minor transplantation antigen (108). More recent protocols now combine DST with other tolerance-inducing methodologies. In addition, it has also been demonstrated that cells other than small B cells can mediate the tolerizing effect of DST. These protocols are discussed in section IVA10. 3. Peptide-based therapy Donor-specific transfusion is a complex biologic process, rich in both peptide antigens and APC. To investigate the antigenic specificity of the donor antigen that is important in the induction of tolerance, some investigators have focused on peptide-induced hyporesponsiveness. The use of peptides rather than DST permits focused investigation of the role of indirect antigen presentation in allorecognition, since by design, the donor antigen cannot be presented by the direct presentation pathway by donor APC (24). Peptides of MHC class I and class II antigens have been used to successfully induce tolerance when administered to recipients orally (347) or with cyclosporine A (71). 4. Intrathymic injection of antigen Injection of soluble antigen and cellular alloantigen injection (427) into the thymus to induce tolerance was first demonstrated in the 1960s. The implications of this / 9j0d$$ja01 P26-8 119 observation were not fully appreciated until much later. This tolerance induction method was first applied to a clinically relevant model of islet transplantation in 1990 (311). Intrathymic injection of allogeneic islets in combination with a single dose of antilymphocyte serum to delete preexisting alloreactive peripheral T cells led to permanent restoration of normoglycemia in a chemically diabetic recipient. This paradigm of tolerance induction based on the intrathymic injection of alloantigen (in most cases, intact donor cells) plus deletion of preexisting T cells was confirmed for islets (50, 250, 310, 311) and extended to other tissues, including skin (84, 286), heart (121, 281–283, 367), liver (49), and kidney (325). The protocol for intrathymic injection of alloantigen was further extended to include intrathymic injection of peptides (287, 348) and soluble antigens (288). As was also true of the peripheral peptide injection protocols that succeeded DST, any tolerogenic effect of soluble antigen injected intrathymically would have to depend on the indirect pathway of antigen presentation. Induction of tolerance using intrathymic peptides appeared to be mediated by the induction of T-cell anergy because the tolerant state could be broken by administration of exogenous IL2 (26, 235). In contrast, it has been suggested that the tolerance induced by the intrathymic injection of spleen cells results from clonal deletion of alloantigen-reactive thymocytes (160). Deletion of antigen-specific thymocytes following intrathymic injection of donor cells was documented using TCR transgenic mice that were then able to tolerate a heart allograft. Another study suggested that either clonal deletion or clonal anergy could be important in the tolerance induced by intrathymic injection of alloantigen and that the thromboxane A2 receptor was involved in the process (324). The basic intrathymic injection protocol has been extended by the application of a novel antigen delivery system. Intrathymic injection of DNA has reportedly resulted in the induction of donor-specific tolerance (176). Interestingly, both clonal deletion and peripheral suppression, the latter on the basis of adoptive transfer studies, were implicated in the tolerance inducted by this protocol. In summary, intrathymic injection of alloantigen in combination with depletion of preexisting alloreactive peripheral T cells can induce permanent tolerance to a number of tissues and may be clinically applicable. The protocol has not yet, however, been reported to work in larger animals or primates. In addition, the progressive involution of the thymus that occurs as humans age poses at least a theoretical obstacle to this approach in the older individuals most likely to require a transplanted organ. 5. Oral antigen administration Oral tolerance has been used successfully for the treatment of autoimmune disorders in many animal mod- 12-09-98 00:40:31 pra APS-Phys Rev 120 ROSSINI, GREINER, AND MORDES els (437–439) and for the induction of tolerance to soluble antigens (61, 113). Its efficacy in the treatment of human autoimmunity remains uncertain, however, and there is no evidence to date in any species that it can induce allotolerance and prolong allograft survival. 6. Monoclonal antibody therapies Monoclonal antibodies directed against cell-surface molecules important in immune activation and response can be used to manipulate the immune system. This approach is attractive for several reasons. Monoclonal reagents have consistent effects in multiple individuals. They target only a single epitope on a single target molecule. Finally, the surface molecules on cells of the immune system tend to be present across species, making a monoclonal antibody-based therapy developed in a rodent model likely to be applicable to primates. We next briefly consider the monoclonal antibodies that have been found to modulate responses important in transplantation. A) ANTI-CD4. Antigen administered in combination with anti-CD4 MAb can induce tolerance to that antigen (78, 341, 368, 369). Several mechanisms have been hypothesized to account for this form of tolerance induction. The first anti-CD4 MAb studied were depleting antibodies, and it was suggested that CD4/ helper/inducer alloreactive cells were simply deleted, resulting in the inability of the host to respond to the foreign graft (7, 365, 368, 378). The depleted cells were shown to be naive CD4/ cells, suggesting that depleting anti-CD4 MAb treatment affects non-antigen-experienced effector cells (326). It was later shown, however, that tolerance could also be induced using nondepleting anti-CD4 MAb, requiring new hypotheses to account for the observed results (76, 370). Two potential mechanisms have been studied extensively: immune deviation (28, 78, 186, 227, 320, 370) and induction of suppressor cell activity (211, 246, 292, 362). The term infectious tolerance (see section IIIE3D) has been used to define the suppressor cell activity that is induced by anti-CD4 MAb treatment. It implies that the cells of the tolerant host can inhibit the response of normal T cells to the foreign graft (292, 314). Anergy induction may also play a role in anti-CD4 MAb-induced suppression, based on the demonstration that the tolerant state can be broken by the addition of IL-2 (6, 58). Other investigators, however, have failed to find evidence of anergy following tolerance induction with five different clones of anti-CD4 MAb (309). This observation raises the possibility that the anergy may be involved only in a subset of cases of anti-CD4 MAb-induced tolerance. It has also been suggested that anti-CD4 MAb can induce Fas-mediated apoptosis of CD4/ T cells (19, 431). This mechanism for the induction of tolerance by antiCD4 MAb has only recently been suggested, and its potential importance is uncertain. / 9j0d$$ja01 P26-8 Volume 79 Regardless of the mechanism involved, anti-CD4 MAb treatment has been shown to induce long-term survival of islet and heart allografts in mice (6, 369) and rats (211). Its potential as an adjunctive therapy in clinical transplantation remains to be evaluated. B) ANTI-CD45. The CD45 family of transmembrane protein-tyrosine phosphatases plays a critical role in T-cell signaling (55, 419, 451). Multiple alternatively spliced CD45 isoforms, differing only in their extracellular domains, are differentially expressed by subsets of T cells with distinct functional repertoires. Very soon after the identification of CD45, it was observed that antibodies to CD45 could inhibit mixed lymphocyte reactions in vitro (141, 205). With the extension of these findings, it was observed that antibody to CD45RB could delay renal allograft rejection in mice (206). Attempts to replicate this observation with other clones of anti-CD45RB antibodies were not successful, however. Initially, only antibody from clone MB23G2 could mediate the effect, but it has now been found that anti-CD45RB produced by multiple hybridoma clones can prolong islet allograft survival (13). The mechanism by which anti-CD45RB antibody induces tolerance is not known. Proposed mechanisms include differential intracellular signaling regulating IL-2 production and activation of signal pathways involving Vav in T cells (251, 291). It has also been shown that distinct isoforms of CD45 can differentially regulate TCR-mediated signals (141, 251, 291). The differential regulation of Tcell activation signals may prevent the generation of an alloimmune response or may induce other forms of tolerance based on suppression, anergy, or clonal deletion. C) ANTI-LYMPHOCYTE FUNCTION ANTIGEN AND ANTI-INTRACELLULAR ADHESION MOLECULE-1. Antibodies to the adhesion molecules lymphocyte function antigen (LFA) and intracellular adhesion molecule-1 (ICAM-1) have been shown to induce permanent heart allograft (156) and prolonged skin allograft (157) survival in mice. Anti-LFA and anti-ICAM antibodies can also prolong rat-to-mouse cardiac xenograft survival (256) and human-to-mouse islet xenograft survival (456). These antibodies are thought to mediate their graft-prolongation effects not by tolerance induction, but rather by inhibition of the migration of host immune cells into the graft. Anti-LFA MAb has also been used to prolong survival of transplanted myoblasts, but in this report, host infiltration into the graft was observed, suggesting that other mechanisms must be important (132). D) ANTI-MHC CLASS I. Human pancreatic islet xenografts have been successfully transplanted into mice without immunosuppression after coating of the donor islets with F(ab*)2 fragments of anti-HLA class I antibody or antibody to tissue-specific epitopes (94). Whether tolerance is induced using this approach or whether the host immune system simply fails to recognize the grafted tissue is not known. 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION E) ANTI-MHC CLASS II. Administration of recipient-specific anti-MHC class II antibodies has been shown to prolong heart allograft survival in rodents (313, 376). The anti-MHC class II antibody does not have to be cytotoxic, suggesting that it is working via immune modulation, not simply the depletion of activated class II/ rat T cells (376). Anti-MHC class II antibody does not, however, prolong skin allografts. It has been hypothesized that the effect of antirecipient MHC class II antibody is mediated by redirection of host immune responses toward a suppressor phenotype (38, 443). F) IN VITRO CULTURE TO REMOVE PASSENGER LEUKOCYTES. Donor APC present in a graft are important mediators of host immune responses via the direct antigen presentation pathway. Recognition of this important role of graft APC led to the discovery that their removal could have important beneficial effects on allograft survival (134, 389). These beneficial effects on graft survival are hypothesized to be due to the inability of the graft to present antigen and to sensitize the recipient host immune system. Graft APC have been removed from islet (95) and kidney (207) allografts by pretreatment with antibody to remove MHC class II-expressing cells. Other approaches to remove donor graft APC include dietary manipulation (210, 354) and, for islets, in vitro culture (245, 411). 8. Interference with costimulation Recognition that costimulation signals are required to achieve full T-cell activation after TCR ligation with MHC plus antigen has led to several tolerance induction strategies based on blockade of this interaction. The fusion protein CTLA4-Ig, which effectively blocks costimulation via CD28 by binding to B7–1/2 on APC, prolongs kidney (14, 345), heart (300, 302, 422, 460), and islet (54, 418) allograft survival. It also prolongs the survival of xenogeneic human islets in mice (214). Treatment of islet allografts with CTLA4-Ig also prolongs their subsequent survival in mice, presumably by inhibiting the ability of passenger leukocytes to provide direct antigen presentation and costimulation (390). The mechanisms hypothesized to be involved in tolerance induction following administration of CTLA4-Ig include suppression and immune deviation resulting from selective inhibition of Th1-type immune responses (305, 345, 418). Treatment with CTLA4-Ig can, however, prolong heart allograft survival in IL-4 knockout recipients, suggesting that the generation of a Th2-type immune response is not necessary for tolerance induction with this reagent (194). 9. Interference with coactivation As outlined in section IIIE1, coactivation describes the interaction of CD40 on APC with its ligand CD154 on T cells; it is an important step that precedes the induction / 9j0d$$ja01 P26-8 121 of costimulation (66, 103). Blockade of coactivation in the presence of a graft would therefore be predicted to lead to the presentation of antigen in the absence of costimulation. This hypothesis has been tested experimentally. The first studies using antibody to block coactivation in transplantation examined an islet transplant model (297, 337). Transplantation of islet allografts into mice treated with anti-CD154 MAb resulted in permanent survival of Ç40% of the grafts. In contrast, administration of anti-CD154 MAb did not prolong skin allografts (244). The same observation was made for rat to mouse xenografts (118, 240). The reason for the prolongation of islet allografts, but not skin allografts or xenografts was not clear. It could have resulted from the large number of Langerhans cells present in skin; such cells provide costimulatory activity much more efficiently than do cells present in islets. Alternatively, the effect could have stemmed from the induction of costimulatory molecules on donor graft APC due to tissue damage and/or nonspecific inflammatory processes not directly dependent on CD40-CD154 interactions (244, 297, 337). 10. Combination methodologies Combinations of immunosuppressive agents are superior to monotherapy in the prevention of acute graft rejection. Similarly, the broad diversity of approaches to tolerance induction, no one of which is uniformly successful as monotherapy for all grafts in all species, has led to the evaluation of combination protocols. The goal has been to join strategies that would act synergistically in the induction of transplantation tolerance. Combination therapies for the prevention of acute graft rejection fall into two general categories. The first combines immunosuppressive drugs with immune modulation. The second combines two or more immunomodulatory approaches that target different elements of the immune system that participate in graft rejection. In this section, we first describe the combined immunomodulatory approach our own laboratory has used to induce tolerance to islet and skin allo- and xenografts. We then compare and contrast it with combination immunomodulatory therapies that have been used by other investigators. A) COMBINATION OF DST AND ANTI-CD154 MAB: A TWO-ELEMENT PROTOCOL FOR TOLERANCE INDUCTION. Based on the apparent requirement for an active immune response for induction of durable tolerance, investigators from our laboratory have focused on a combination approach using two tolerance-inducing methods: the administration of DST to induce an active immune response and the blocking of CD40 and CD154 coactivation to induce tolerance in the activated alloreactive cells. We describe this as a two-element protocol. Our approach to the induction of tolerance targets 12-09-98 00:40:31 pra APS-Phys Rev 122 ROSSINI, GREINER, AND MORDES early events that accompany T-cell activation, events that occur immediately after ligation of the TCR with MHC plus peptide. It is directed specifically at the blockade of coactivation by interfering with the interaction of molecules required for this process. Blockade of costimulation inhibits the subsequent induction of B7–1/2 costimulatory molecules on APC. In our first experiments, we used a population of small lymphocytes enriched for resting B cells as APC to present alloantigen in the absence of coactivation signals (297). Small B lymphocytes express low levels of B7–1/2 on their surface and are ineffective APC (45, 116, 124, 158, 296) (see sect. IVA2). As described in section IIIE1, T cell-dependent activation of resting B cells leading to the expression of B7–1/ 2 costimulatory molecules depends largely on the interaction of CD154 on T cells with its receptor, CD40, on B cells (197, 277, 295, 381). CD154 on T cells is expressed primarily by activated CD4/ T cells; it is not expressed on resting T cells and is expressed only at low levels on activated CD8/ T cells (277, 295). The engagement of CD40 with its ligand, CD154, results in the rapid upregulation of B7–1/2 on B cells and other APC (168, 319). To induce tolerance, we blocked this interaction using the MR1 hamster anti-mouse CD154 MAb. This reagent prevents cognate T- and B-cell signaling and inhibits T-dependent humoral responses in vitro and in vivo (112, 278). Anti-CD154 MAb prevents expression of autoimmunity in several experimental models (66, 77, 103, 122, 195). These include experimental autoimmune arthritis (89), experimental allergic encephalomyelitis (114, 123, 344), thyroiditis (51), lupus in murine models (72, 90, 304), and spontaneous diabetes in nonobese diabetic (NOD) mice (18). In addition, anti-CD154 MAb impairs allo-specific CTL responses in vitro, prevents the occurrence of acute and chronic GVHD following injection of allogeneic bone marrow (88). It also promotes the generation of allogeneic hematopoietic chimeras and permanent transplantation tolerance in the T-cell compartment when combined with low-dose irradiation (33, 440). Given these data, we developed a two-element protocol consisting of a pretransplant infusion of donor-specific splenocytes enriched for resting B cells plus a course of anti-CD154 MAb injections. We hypothesized that the protocol would induce permanent allograft survival because the DST would initiate T-cell activation (signal 1), but in the absence of costimulation (signal 2). Absence of costimulation, which had been blocked by interference with CD154-dependent coactivation signals, would induce T-cell nonresponsiveness to the donor alloantigen in the host (Fig. 3). The theory predicted that the actual treatment period could be short, needing to be applied only during the immediate peritransplant period. The theory also predicted that the tolerance induced should be durable be- / 9j0d$$ja01 P26-8 Volume 79 cause most or all alloreactive T cells should become unresponsive. This protocol was first tested in an islet allograft model. Administration of a single DST and a 2-wk course of anti-CD154 MAb (totaling 4 injections) induced permanent islet allograft survival in ú95% of chemically diabetic hosts (297, 337). This result suggested that once tolerance to the allograft was induced, further modulation of the immune system may not be necessary. We then extended the evaluation of this protocol to determine if tolerance could be induced to skin allografts. Because of the robust antigenicity of skin compared with islets, it affords a much more challenging assay of transplantation tolerance. Our two-element protocol prolonged survival of murine skin allografts. Median skin graft survival time was Ç50 days, and 20% of skin grafts survived ú100 days (244). Most recently, we have shown that the protocol can be effective when applied to concordant rat to mouse xenografts. In preliminary studies, combination therapy with DST and anti-CD154 MAb significantly prolonged islet xenograft survival (240). Skin xenograft survival was not prolonged (240). Subsequent refinement of the protocol has, however, lengthened skin xenograft survival significantly, to a median of 36 days (118). These encouraging results have engendered intense investigation of the two-element approach to induction of transplantation tolerance using anti-CD154 MAb in combination with DST. An alternative approach using antiCD154 MAb in combination with simultaneous blockade of CD28 and B7–1/2 costimulation is also being extensively investigated (see sect. IVA10C). The promising outcomes achieved with DST and antiCD154 MAb have already generated talk of clinical trials. The available data, however, leave unanswered at least four important questions that should be addressed before translation of this protocol to the clinic can be considered. First, which component(s) of the host immune response is being modulated, and can the tolerance-induction protocol be improved by targeting those components more effectively? Second, what mechanism(s) is involved in the induction and maintenance of the induced tolerance? Third, how durable is the tolerant state? Can it be broken easily by environmental perturbants? These questions can be addressed in rodents where the model system can be easily manipulated. Finally, will this two-element protocol work in larger animals, specifically primates and humans? Recent studies have provided insights, and in some cases answers to most of these questions. i) Affected components of the immune system. Extensive investigations have been carried out regarding the components of the host response affected by this twoelement protocol, and these have led to its improvement. The effects of anti-CD154 on the immune system per se appear to be primarily directed at CD4/ T cells. Antibody treatment can prevent the coactivation of helper/inducer 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION CD4/ T cells and most of the effector and helper T- and B-cell responses that are dependent on this cell subset (66, 103, 195). In allograft transplantation, this effect stems primarily from the blockade of T cell-APC interaction via CD40-CD154 and prevention of the subsequent upregulation of B7–1/2 costimulatory molecules on APC. Other activities of anti-CD154 MAb that may be important in prevention of transplant rejection include effects on NK cells (52), macrophages and dendritic cells (31, 247, 395), and endothelial cells (166, 234, 425). It has also been shown that activated platelets express CD154 (148). The role of rapid upregulation of CD154 on activated platelets leading to endothelial cell activation has been shown to occur in vitro and in vivo. The ability of anti-CD154 MAb to block the coactivation activity of platelet-expressed CD154 on endothelial cells may be responsible, in part, for nonantigen-specific anti-inflammatory effects that are observed after administration of anti-CD154 MAb (148). Refinement of the protocol has yielded exciting information on the mechanism(s) involved and the durability and stability of the tolerance that is induced. Cell composition of the DST has shown that small B cells are sufficient, but not required, for tolerance induction. Administration of whole spleen cell preparations is efficacious (244), as is administration of spleen cells from B-lymphocyte knockout mice. T cells are not required in the donor transfusion, because administration of spleen cell preparations from scid mice also induces tolerance in combination with anti-CD154 MAb. Finally, cell fractions enriched for dendritic cells can function as the cell population as the DST in combination with anti-CD154 MAb (229, 242a, 440). We conclude that, in combination with anti-CD154 MAb, a broad range of cell types can be used for the DST component of our two-element protocol. Experiments that modify the host immune response have also yielded exciting insights into the basis for the late failure of skin allografts following tolerance induction using our two-element protocol. Thymectomy of the host before starting the protocol results in permanent skin allograft survival in ú95% of recipients (243), an outcome similar to that achieved in euthymic mice given islet allografts (297). These data suggest that the short-term treatment protocol induces tolerance in the preexisting T-cell population but fails to induce tolerance in thymic emigrant populations that emerge later. This interpretation predicts that modifications of the protocol that either periodically tolerize new thymic emigrants or prevent their emergence from the thymus will enhance the durability of induced graft tolerance. ii) Mechanism. Recent experiments have also provided some insight into the mechanism(s) involved in the induction and maintenance of tolerance. The role of CD4/ cells in both the induction and maintenance phases of the graft-host interaction is now clear. Administration of antiCD4 MAb but not anti-CD8 MAb before starting the two- / 9j0d$$ja01 P26-8 123 element protocol abrogates tolerance induction (243). This result contrasts sharply with previous reports that graft survival can be prolonged using only anti-CD4 MAb (see sect. IVA6A). Our result suggests that an active process, mediated by CD4/ T cells, is critical for tolerance induction by the two-element protocol. Furthermore, administration of depleting anti-CD4 MAb to mice that have had intact skin grafts for long periods of time (ú100 days) results in rapid graft rejection (243). These effects could be mediated by the elimination of CD4/ suppressor cells or by alteration of the cytokine profile of the remaining cells, i.e., immune deviation. However, DST and anti-CD154 MAb readily induce tolerance in IL-4 knockout mice, as well as in normal and thymectomized mice that have been treated continuously with antiIL-4 MAb, a Th2-type cytokine. Tolerance cannot be induced by the two-element protocol in mice treated with anti-IFN-g or in IFN-g knockout mice, hosts strongly biased toward Th2-type immune responses. These data suggest that if active suppression is responsible for tolerance induction, it does not occur through the mechanism of immune deviation (243). Finally, CD28 and B7–1/2 interaction also appears to play a critical role during the early phases of tolerance induction when our protocol is applied. Administration of anti-CTLA4 MAb prevents the induction of tolerance (243). This observation suggests that interaction of CDTL4 with its ligands B7–1 and B7–2 is essential in the tolerance induction process. This last result contrasts sharply with reports of investigations on the effect of combination therapy consisting of anti-CD154 MAb and CTLA4-Ig. Administration of both reagents reportedly induces tolerance to skin and heart allografts in mice even in the absence of DST (201). iii) Induction and maintenance phases. Taken together, these observations have led to the hypothesis that the induction of tolerance using DST plus anti-CD154 MAb involves at least two distinct critical stages or phases (see Table 1). Phase 1 is the induction phase. This occurs during administration of the DST and the anti-CD154 MAb. Phase 1 requires CD4/ T-cell activation, depends on the interaction of CTLA4 with its ligands B7–1 and B7–2, and requires IFN-g. The second clearly delineated phase involves the maintenance of tolerance in recipients with long-term intact grafts. This phase is also dependent on the presence of CD4/, presumably suppressor T cells. Our hypothesis predicts that the relative balance of CD4/ suppressor cells with alloreactive recent thymic emigrants ultimately determines the long-term fate of the graft. The ‘‘two-phase’’ or ‘‘two-stage’’ concept of tolerance induction predicts that the first phase, induction, may be relatively unstable. It appears to be dependent on the administration of DST and the continued presence of antiCD154 MAb. The induction phase may easily be broken 12-09-98 00:40:31 pra APS-Phys Rev 124 ROSSINI, GREINER, AND MORDES TABLE Volume 79 1. Phases of tolerance induction by donor-specific transfusion and anti-CD154 monoclonal antibody Phase Induction Treatments Immunologic events Tolerance Transplantation Transition Transfusion of alloantigen; anti-CD154 MAb begins IFN-g required; CD4/ T cells required; CTLA4 ligation required Surgery; anti-CD154 MAb continues Second exposure to alloantigen; surgeryinduced inflammation; graft antigenicity: islets low, skin high ‘‘Tolerization’’ of preexisting peripheral T cells begins ‘‘Tolerization’’ of preexisting peripheral T cells continues; induction of CD4/ suppressor cells Permanence Anti-CD154 MAb ends No treatments Graft susceptible to rejection due to alloreactive recent thymic emigrants; in vitro graft alloreactivity demonstrable; CD4/ T cells required; graft susceptible to rejection due to environmental perturbants Metastable equilibrium between ‘‘untolerized’’ new alloreactive thymic emigrants and CD4/ suppressor cells CD4/ T cells required; minimal in vitro graft alloreactivity demonstrable Recognition of graft as self MAb, monoclonal antibody; IFN-g, interferon-g. by introduction of environmental perturbants while antiCD154 MAb is still being administered. This inference is supported by our observations 1) that altering the timing of the DST or anti-CD154 MAb relative to graft placement influences graft survival and 2) that extended administration of anti-CD154 MAb is associated with prolonged skin allograft survival (242). Our hypothesis also predicts that once tolerance is induced, it is relatively stable. This is evidenced by the long-term survival of both islet and skin grafts that have not undergone acute rejection during the induction phase. Our concept of the stages of allotolerance induction that follow treatment with DST and antiCD154 MAb is depicted schematically in Figure 4. iv) Durability. We hypothesize that the durability of transplantation tolerance may depend on events that occur during the transition from the induction to maintenance phases of the host response (193). We propose that there may exist a third ‘‘metastable’’ phase of tolerance. The metastability of this phase would explain the loss of grafts that are intact during the actual treatment and escape acute rejection, but are nonetheless eventually rejected. We propose that the metastable phase is dynamic, of varying duration, and dependent on active immune responses. Conceptually, it may in part depend on either 1) the relative balance of suppressor and alloreactive effector cells that remain after the induction protocol is complete or 2) the microenvironmental cytokine milieu in the graft or the systemic circulation of the host. The ability to transit the metastable phase to the long-term maintenance phase may also depend on 1) the degree of inflammation induced in the host due to surgical manipulation during transplantation and 2) the resilience of the graft as it recovers from the surgical and inflammatory trauma of transplantation. B) COMBINATION OF CTLA4-IG AND DST. CTLA4-Ig has as / 9j0d$$ja01 P26-8 strong affinity for, and ability to block, the interaction of the B7–1/2 costimulatory molecule with CD28. These characteristics predicted it to be a potent immunoregulatory of immune responses important in allograft rejection. It was observed in early studies of CTLA4-Ig fusion protein in transplantation that the CTLA4-Ig immunomodulatory agent promoted graft survival more efficiently if combined with exposure to alloantigen by DST (345, 350). Administration of CTLA4-Ig by itself was insufficient to prevent heart allograft rejection, but when combined with DST, vascularized heart allograft survival was prolonged significantly (218). Administration of CTLA4-Ig plus bone marrow also prolonged the survival of cardiac allografts in mice receiving a completely MHC-mismatched graft, but the treatment did not prolong skin allograft survival (301). Surprisingly, the cytokines detectable in graft recipients that were treated with CTLA4-Ig and DST were identical to those detected in unmanipulated control graft recipients. The expression of granzyme, perforin, Fas, and FasL mRNA was also similar in treated and control graft recipients. The experiments also revealed that Th2-type cytokines were readily detectable in the hosts and that CTLA4Ig appeared specifically to inhibit the Th1-type T-cell immune response. The data suggest that graft survival in animals treated with CTLA4-Ig plus DST may involve multiple mechanisms, including microchimerism, anergy, and immune deviation (301). C) COMBINATION OF CTLA4-IG AND ANTI-CD154 ANTIBODY. A second two-element protocol consisting of anti-CD154 MAb and CTLA4-Ig has also been evaluated for transplantation tolerance induction (201). This approach is based on the hypothesis that anti-CD154 MAb should prevent interaction with CD40 and coactivation, while at the same time CTLA4-Ig should prevent costimulation of any 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION 125 FIG. 4. Depicted in schematic form is our concept of stages of allotolerance induction that occur during and after treatment with donor-specific transfusion (DST) and anti-CD154 monoclonal antibody (MAb). During treatment, suppressor cell populations are induced, and alloreactive T-cell activity begins to decline. When graft is actually transplanted at conclusion of this ‘‘induction’’ phase, alloreactive cell activity declines greatly. After transplantation, there occurs a transition period of variable duration during which time graft is ‘‘metastable.’’ This is depicted as a balance between alloreactive (A) and suppressor (CD4) cells that may be disequilibrated in either direction. If a balance progressively favoring suppressor cells is maintained through transition period, then graft survival (depicted here as a mouse skin graft typical of those on which this concept is based) becomes permanent. IFN-g, interferon-g. alloreactive T cells that were either preactivated or escaped the effects of CD40-CD154 blockade. The combination of these two reagents prolongs the survival of fully allogeneic skin and vascularized heart allografts. In skin graft studies, it was shown that both reagents were required; neither was effective by itself. In vascularized heart allografts, anti-CD154 MAb by itself was able to prolong graft survival (200). Combined treatment with antiCD154 MAb and CTLA4-Ig was also able to prevent the chronic rejection commonly associated with mouse aortic allografts (399). The mechanism(s) directly responsible for the tolerance induced by this approach have not been identified but are likely to involve both redundant blocking of both coactivation and costimulation as well as additional factors (351). the acute rejection of allografts and facilitate their longterm survival. Any transplant protocol in humans that excludes generalized immunosuppression, however, must be robust and durable as documented in primate studies. Unfortunately, the application of tolerance induction protocols pioneered in rodents to larger animals, specifically primates, has proven difficult. As in rodents, there have been two general experimental approaches to the induction of transplantation tolerance in primates. The first is the use of immunomodulatory reagents in the absence of general immunosuppression; the second is the use of general immunosuppression with or without immunomodulatory reagents. 1. Anti-CD154 MAb and DST in large animal models D) CTLA4-IG PLUS OTHER IMMUNOMODULATORY OR IMMUNOSUPPRESSIVE AGENTS. Experiments featuring the combination of CTLA4-Ig with other drugs have been undertaken in part because of the belief that utilization of any new agent in clinical transplantation will almost certainly require its combination with currently used agents. Accordingly, CTLA4-Ig has been tested in combination with cyclosporine (39) and tacrolimus, two conventional immunosuppressive agents (136). Although prolongation of graft survival was observed, graft rejection did eventually occur in all cases. Finally, CTLA4-Ig has been combined with the promising technique of intrathymic injection of alloantigen (382). Again, graft survival in this experiment was prolonged but not permanent. B. Methods Successful in Large Animals and Primates Data from rodent experiments suggest that there are multiple avenues for immunomodulation that may prevent / 9j0d$$ja01 P26-8 Among species, CD40 and CD154, as well as CD28 and B7–1/2, are strongly conserved. The coactivation and costimulation pathways mediated by these molecules appear to be important in the regulation of immune responses in rodents, larger mammals, primates, and humans (66, 103, 195). The possibility therefore exists that the basic paradigm developed in murine models may be extendable to primates and perhaps even to humans. Preliminary indications suggest that this may, in fact, be the case. In studies of primates, two-element therapy composed of DST and anti-CD154 MAb prolongs kidney allograft survival (336). Administration of anti-CD154 MAb, with or without the addition of CTLA4-Ig to block CD28 and B7–1/2 interactions, also can prolong kidney allografts in primates (see sects. IVA9 and IVA10C). The preliminary results in rodents and primates now suggest that clinical trials using the two-element protocol may be feasible. The development of anti-human CD154 12-09-98 00:40:31 pra APS-Phys Rev 126 ROSSINI, GREINER, AND MORDES MAb, and the humanization of these MAb (104, 171), should permit this approach to be extended to the clinic in the near future. 2. Anti-CD154 MAb and CTLA4-Ig in large animal models Several published reports on tolerance induction in monkeys using immunomodulatory reagents without immunosuppression have employed CTLA4-Ig alone or with anti-CD154 MAb (171, 215). Using CTLA4-Ig alone, two of five monkeys with chemical diabetes that were treated with CTLA4-Ig and transplanted with allogeneic islets became normoglycemic and remained free of diabetes for 30 and 50 days without additional treatment (215). In a second study, rhesus monkeys were transplanted with allogeneic kidneys and given human CTLA4-Ig, humanized anti-human CD154 MAb, or both reagents (171). In these monkeys, either reagent alone prolonged graft survival to 20–98 days, and in two of the four monkeys treated with both reagents, kidney allograft survival was ú150 days. In the absence of continuous treatment however, episodes of rejection occurred; in these instances, administration of additional anti-CD154 MAb preserved graft function. Other preliminary reports in primates describe prolonged survival of islet allografts in pancreatectomized cynomologus monkeys, baboons, and rhesus monkeys treated with anti-CD154 MAb alone (169, 212). In these cases, prolonged graft survival required a brief induction course of anti-CD154 MAb in the perioperative period and additional monthly maintenance doses. In all of these cases in which antibody alone was used to induce transplantation tolerance, it will be of interest to determine if passenger APC in the donated graft subserved the function of a DST. These results, combined with our preliminary experience using the two-element protocol in monkeys, suggest that immunomodulatory reagents targeted to the coactivation and costimulation molecules on T cells and APC have the potential to induce transplantation tolerance in humans. 3. Other immunomodulatory approaches in large animal models Several other approaches have been employed using various forms of generalized or specific immunosuppression. These include the use of the immunotoxin FN18CRM9, which is an anti-rhesus CD3 MAb (FN18) conjugated to the mutant diphtheria toxin protein CRM9 (271). This reagent has been used to induce tolerance in monkeys to kidney (96) and skin (175) allografts in combination with intrathymic injection of donor lymphocytes. The combined protocol appeared to downregulate antidonor CTL activity in the kidney allograft recipients, whereas / 9j0d$$ja01 P26-8 Volume 79 helper T-cell and B-cell function and reactivity to third party allografts remained unaffected. A more traditional approach has employed combinations of various immunosuppressive techniques for tolerance induction to kidney allografts in cynomologus monkeys (170a). The preparative regimen included various combinations of whole body low-dose irradiation, antithymocyte globulin, thymic irradiation, splenectomy, and donor bone marrow infusion with or without a short course of cyclosporine. The conclusion drawn from this study was that long-term graft survival can be obtained using combination immunosuppressive therapy, but the best predictor of graft survival was the extent of chimerism achieved. These limited studies using only immunomodulatory therapy or low-dose immunosuppressive regimens with immunomodulatory agents have provided promising evidence that it will prove possible to induce transplantation tolerance in primates. However, the mechanism(s) responsible for graft survival in these primate protocols remains unknown and will be difficult to analyze experimentally. The combination of rodent experimentation and extrapolation to the primate offers the most promising approach to the development of optimal tolerance induction protocols with applicability to humans. C. Evidence of Tolerance Induction in Humans Because of the universal acceptance of immunosuppression in current clinical practice, there is little evidence to document the presence or absence of immunologic tolerance in human graft recipients. Among the few available data are those reported by Starzl et al. (385). Those data focus on human graft recipients who for various reasons discontinued immunosuppressive therapy but nonetheless continued to retain their allograft (385). Those data were interpreted to suggest that passenger leukocytes migrate after organ transplantation and produce a microchimeric state that is essential, and in some cases sufficient, for allograft maintenance. One other set of observations is also consistent with the existence of a tolerant state. These data are from human liver allograft recipients and suggest that this tissue may have the ability to produce a tolerant state based on the uniquely high antigenicity of this organ (385). V. CONTEMPORARY CLINICAL ORGAN TRANSPLANTATION A successful organ transplantation program must establish a set of outcome criteria that must be met and must identify and overcome obstacles to meeting those criteria. All criteria are, of course, based on the premise that good donor tissue is available. Mundane but crucial 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION methodologies must be perfected. These include organ and tissue harvesting techniques, preservative solutions, and preparative manipulation, and protocols unique for each tissue or organ have been formulated. A. Criteria for Successful Transplantation Program The first criterion is availability of organs or tissues for transplantation. The recovery and distribution of human organs and tissues for transplantation has been organized nationally under a program titled UNOS (United Network for Organ Sharing). This scientific registry reported in 1995 that its organ recipient waiting list had grown to Ç38,000 individuals, and it is estimated that 9 persons die each day awaiting a human allograft. The shortage of organs for transplantation continues to grow. The second criterion is viability of the organ or tissue to be transplanted. Each tissue or organ again has its own unique requirements, but grafts can be grouped into two general categories. One group includes vascularized organs that require surgical anastomosis of the recipient’s vascular system. The second group includes nonvascularized tissues. These require transplantation into a site where they can be bathed in tissue fluids that are adequate to furnish nutrients and remove or dilute waste metabolites. The first group includes heart, kidney, liver, and lung. The second group includes islets of Langerhans. This dispersed tissue that can be implanted intraportally or intraperitoneally where they survive in the absence of surgical attachment to the vascular system. The third criterion is functionality of the transplanted organ or tissue. Simple survival of the transplant is obviously not the critical outcome variable. The transplanted organ must replicate the function of the organ that it is replacing. The fourth criterion is application of appropriate harvesting techniques. Clearly, harvesting of solid organs requires different approaches than does harvesting of tissues like islets. All approaches have in common a minimization of ischemia time and the use of preservative solutions, with each preservative solution tailored to the particular organ or tissue. For example, to recover islets, the pancreas must be perfused immediately with specific preservative solutions such as Wisconsin’s solution. The islets must then be dissociated from the pancreas within hours of harvest, usually by collagenase digestion, cultured for several days, and if not transplanted at the end of culture cryopreserved for transplantation in the future (188, 189). For each organ or tissue, the optimal protocol is detailed and idiosyncratic. The fifth criterion is safety of the donor organ or tissue. Safety preeminently focuses on bacterial and viral infection of the donor host organ or tissue. Donors with / 9j0d$$ja01 P26-8 127 bacterial septicemia may not be appropriate donors. Donors infected with hepatitis virus, human immunodeficiency virus, or other pathogenic viruses are not acceptable donors. All transplant programs screen for a number of viruses and bacterial pathogens before use of the donor tissue for transplantation, but in no cases can the surgeon be completely ensured of the pathogen-free status of the donor organ or tissue. The special issue of infected xenografts is considered in section VID. B. Issues of Graft Placement The site of transplantation may be critically important for the survival and function of a graft. For many grafts, this requirement constrains choices for the site of graft placement. The graft must be positioned to allow it to perform the function of the organ or tissue being replaced. For vascularized grafts such as the heart, liver, or lung, the graft must be an orthotopic graft, i.e., placed in the normal position for that tissue or organ. Survival of the graft in an unsuitable anatomic location is of no benefit to the recipient. In contrast, kidneys and insulin-secreting islets can be transplanted heterotopically, i.e., in a site different from the natural location. Implantation of islets into the liver or peritoneum rather than the pancreas not only achieves physiological function but may also enhance the chances of graft survival. C. Immunosuppression Immunosuppression is currently required for the survival of all but autologous and some syngeneic grafts. Although there are many forms of immunosuppression, including general and targeted forms, there is at present no immunosuppressive agent that is completely safe and free of toxic side effects. Immunosuppressive drugs are the current method of choice, but their use restricts transplantation of tissues or organs to life-threatening disorders. An optimal immunosuppressive regimen employed to permit graft acceptance needs to be safe and free of adverse effects to the recipient. The extent and duration of immunosuppression will have a significant impact on the incidence of infections and tumors experienced by the recipient. VI. TRANSPLANTATION AFTER TOLERANCE The focus of this review has been improvement in the safety and efficacy of transplantation by exploiting new insights into immunologic tolerance. The goal is to reduce or eliminate our current dependence on immunosuppressive drugs. It is not clear that tolerance can be induced, and translation of the theories and methods we 12-09-98 00:40:31 pra APS-Phys Rev 128 ROSSINI, GREINER, AND MORDES have reviewed to clinical medicine seems a question of if, not when. We conclude with a brief consideration of some issues that must be confronted as the implementation of tolerance-based therapy proceeds. A. Demand for Transplantation Services Therapy for patients with disorders that were once rapidly fatal have improved dramatically. Hemodialysis for renal failure, afterload reduction for heart failure, and intensive insulin therapy for diabetes (pancreatic islet cell failure) are now routine. As brilliantly successful as these therapies are, however, they are temporary, not curative. As it becomes possible to consider transplantation for individuals who would have been marginal candidates in the immunosuppression era, demand and expectations will certainly rise. 1. Organ transplantation is physiologically curative The availability of organ transplants in the absence of immunosuppression will highlight the physiological advantages of organ replacement over current therapy. One example is in the area of diabetes. Intensive insulin therapy can retard, but not preclude, the life-threatening complications of hyperglycemia. The physiological cost (in terms of hypo- and hyperglycemia, for example) and the financial cost are both high, but in the immunosuppression era, most persons with diabetes take a wait-and-see attitude to islet transplantation. This may change dramatically in an era of tolerance-based therapy for at least three reasons. 1) Long-term safety will be enhanced by the elimination of chronic immunosuppression. 2) Restoration of total islet functionality (e.g., physiological secretion of insulin, proinsulin, C-peptide, glucagon, and somatostatin) is more likely to prevent complications than insulin injection (152). Finally, long-term functionality of transplanted islets may also be able to reverse diabetic complications already present at the time of grafting (99). Volume 79 sues of graft survival and graft function. Very little is as yet known about possible unforeseen consequences of inducing tolerance. Ideally, tolerance would be induced quite specifically to one tissue from one donor, leaving the host immune system’s native capabilities to respond to danger unimpaired. This degree of specificity may not necessarily be achieved, however, and compromises in the ability of a recipient in the areas of infection defense or immune surveillance must be considered. 2. Durability The durability of transplantation tolerance has been a recurring theme in this review. It is not yet known, however, what will be required to maintain graft tolerance for a lifetime or what factors might break tolerance. It remains, for example, to be proven if tolerized grafts will continue to survive if the host is forced to mount an immune response to infection or trauma. 3. Chronic rejection Another major point that confronts any transplant program is the prevention of chronic rejection. The majority of kidney transplants, with a modern day initial acceptance rate of ú90%, inevitably fail due to the development of chronic rejection and the loss of function. The pathophysiology of chronic rejection is poorly understood, and current immunosuppressive therapies often fail to reverse chronic rejection, eventually leading to graft failure. 4. Recurrence of disease It is now recognized that the cure of disease by successful organ transplantation, even with the accompanying risks and side effects of immunosuppression, provides substantial psychological benefit to patients. If tolerance-based transplantation reduces the risk of toxicity and the attendant fear of the procedure, wider recognition of the restoration of spiritual health and wider perception of bodily integrity are likely to increase demands on transplantation centers. Most organs requiring replacement have been lost due to trauma, ischemia, congenital malformation, abnormal metabolic states, or other nonimmune processes. Others, however, have been lost due to autoimmune disease. Examples include lupus nephritis, autoimmune cardiomyopathy, and autoimmune diabetes. In these cases, the autoimmune disease may affect a healthy graft. This is known from studies of monozygotic twins discordant for diabetes. Syngeneic segmental pancreas transplantation from a healthy to a diabetic sibling (which requires no immunosuppression) fails due to recurrence of the original disease in the healthy graft (401). How tolerance-based transplantation will function for patients with underlying autoimmune diatheses is not known. Combination therapies directed at the autoimmunity and at alloantigenicity have been suggested, but how they would interact is also unknown. B. Potential Pitfalls C. Issues of Donor Tissue and Organ Availability 1. Specificity of induced tolerance As tolerance-based transplantation becomes a reality, the inadequate supply of donor tissue will become an ever more serious problem. Cadaveric donor organs are 2. Psychological advantages of organ transplantation Experimental procedures designed to evaluate transplantation tolerance induction of necessity focuses on is- / 9j0d$$ja01 P26-8 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION sufficient for only Ç10% of total need at the present time. The gap between requests for donor tissues and their availability continues to grow. One possible source that remains underutilized is human fetal tissues, but ethical and legal barriers to their use may be insurmountable. A second approach is the development of bioengineered tissues. Bioengineered tissues could take many forms: organs from transgenic and knockout animals, transfected allogeneic cell lines designed to eliminate antigenicity while preserving (or adding) function, and, most speculatively, tissues grown in vitro from pluripotent donor cells (neogenesis). These strategies are all in their infancy and clinically unproven. 2. 3. 4. 5. 6. D. Special Case of Xenografts 7. We have devoted considerable attention in this review to the induction of tolerance to xenografts, focusing on the special immunologic characteristics (e.g., indirect antigen presentation) that distinguish xeno- from allografts. The potential use of xenografts raises special ethical and psychological issues for physicians and recipients (67). More importantly, it raises the specter of introducing new diseases into the human population. From an infectious disease standpoint, the issue is exceedingly complex. The specter of xenozonotic epidemics has been raised and has become a focus of concern (17). 8. 9. 10. 11. 12. E. Conclusion The advances in transplantation biology that have occurred in recent decades are stunning, if not miraculous. Through the use of immunosuppressive drugs, transplantation has become a widespread reality. Potentially safer and more efficacious transplantation methods based on tolerance induction are already a real possibility. One wonders if humans of the future will actually embody the miracle of transplantation to the extent depicted by Fra Angelico in his painting of the patient of Cosmas and Damian. We thank Dr. Jeffrey Stoff for critical review of the manuscript. This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grants DK-25306 (to A. A. Rossini), DK-41235 (to J. P. Mordes), and DK36024 (to D. L. Greiner); by a grant from the Iacocca Foundation; and by Program Project Grant DK-53006, jointly funded by NIDDK and the Juvenile Diabetes Association International (to A. A. Rossini). REFERENCES 14. 15. 16. 17. 18. 19. 20. 1. ABBAS, A. K., A. H. LICHTMAN, AND J. S. POBER (Editors). Immune responses to tissue transplants. In: Cellular and Molecular / 9j0d$$ja01 13. P26-8 21. 12-09-98 00:40:31 129 Immunology (3rd ed.). Philadelphia, PA: Saunders, 1997, p. 364– 381. ACHA-ORBEA, H., A. N. SHAKHOV, L. SCARPELLINO, E. KOLB, V. MULLER, A. VESSAZ-SHAW, R. FUCHS, K. BLOCHLINGER, P. ROLLINI, AND J. BILLOTTE. Clonal deletion of V beta 14-bearing T cells in mice transgenic for mammary tumour virus. Nature 350: 207–211, 1991. AHLSKOG, J. E. Cerebral transplantation for Parkinson’s disease: current progress and future prospects. Mayo Clin. Proc. 68: 578– 591, 1993. ALLEN, J. E., AND R. M. MAIZELS. Th1-Th2: reliable paradigm or dangerous dogma? Immunol. Today 18: 387–392, 1997. ALLISON, J., H. M. GEORGIOU, A. STRASSER, AND D. L. VAUX. Transgenic expression of CD95 ligand on islet beta cells induces a granulocytic infiltration but does not confer immune privilege upon islet allografts. Proc. Natl. Acad. Sci. USA 94: 3943–3947, 1997. ALTERS, S. E., J. A. SHIZURU, J. ACKERMAN, D. GROSSMAN, K. B. SEYDEL, AND C. G. FATHMAN. Anti-CD4 mediates clonal anergy during transplantation tolerance induction. J. Exp. Med. 173: 491–494, 1991. ALTERS, S. E., H. K. SONG, AND C. G. FATHMAN. Evidence that clonal anergy is induced in thymic migrant cells after anti-CD4mediated transplantation tolerance. Transplantation 56: 633–638, 1993. ANTIN, J. H., AND J. L. FERRARA. Cytokine dysregulation and acute graft-versus-host disease. Blood 80: 2964–2968, 1992. ARMSTRONG, H. E., E. M. BOLTON, I. MCMILLAN, S. C. SPENCER, AND J. A. BRADLEY. Prolonged survival of actively enhanced rat renal allografts despite accelerated cellular infiltration and rapid induction of both class I and class II MHC antigens. J. Exp. Med. 165: 891–907, 1987. ARNOLD, B., G. SCHONRICH, AND G. J. HAMMERLING. Multiple levels of peripheral tolerance. Immunol. Today 14: 12–14, 1993. AUCHINCLOSS, H., JR. Xenogeneic transplantation. A review. Transplantation 46: 1–20, 1988. AUCHINCLOSS, H., JR., R. LEE, S. SHEA, J. S. MARKOWITZ, M. J. GRUSBY, AND L. H. GLIMCHER. The role of ‘‘indirect’’ recognition in initiating rejection of skin grafts from major histocompatibility complex class II- deficient mice. Proc. Natl. Acad. Sci. USA 90: 3373–3377, 1993. AUERSVALD, L. A., D. M. ROTHSTEIN, S. C. OLIVEIRA, C. Q. KHUONG, H. ONODERA, A. I. LAZAROVITS, AND G. P. BASADONNA. Indefinite islet allograft survival in mice after a short course of treatment with anti-CD45 monoclonal antibodies. Transplantation 63: 1355–1358, 1997. AZUMA, H., A. CHANDRAKER, K. NADEAU, W. W. HANCOCK, C. B. CARPENTER, N. L. TILNEY, AND M. H. SAYEGH. Blockade of T-cell costimulation prevents development of experimental chronic renal allograft rejection. Proc. Natl. Acad. Sci. USA 93: 12439– 12444, 1996. AZUMA, H., AND N. L. TILNEY. Immune and nonimmune mechanisms of chronic rejection of kidney allografts. J. Heart Lung Transplant. 14, Suppl.: S136–S142, 1995. AZUMA, M., D. ITO, H. YAGITA, K. OKUMURA, J. H. PHILLIPS, L. L. LANIER, AND C. SOMOZA. B70 antigen is a second ligand for CTLA-4 and CD28. Nature 366: 76–79, 1993. BACH, F. H., J. A. FISHMAN, N. DANIELS, J. PROIMOS, B. ANDERSON, C. B. CARPENTER, L. FORROW, S. C. ROBSON, AND H. V. FINEBERG. Uncertainty in xenotransplantation: individual benefit versus collective risk. Nature Med. 4: 141–144, 1998. BALASA, B., T. KRAHL, G. PATSTONE, J. LEE, R. TISCH, H. O. MCDEVITT, AND N. SARVETNICK. CD40 ligand-CD40 interactions are necessary for the initiation of insulitis and diabetes in nonobese diabetic mice. J. Immunol. 159: 4620–4627, 1997. BANDA, N. K., J. BERNIER, D. K. KURAHARA, R. KURRLE, N. HAIGWOOD, R. P. SEKALY, AND T. H. FINKEL. Crosslinking CD4 by human immunodeficiency virus gp120 primes T cells for activation-induced apoptosis. J. Exp. Med. 176: 1099–1106, 1992. BARKER, C. F., AND R. E. BILLINGHAM. Immunologically privileged sites. Adv. Immunol. 25: 1–54, 1977. BARTLETT, S. T., A. NAJI, W. K. SILVERS, AND C. F. BARKER. Influence of culturing on the survival of rat islet allografts and their pra APS-Phys Rev 130 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. ROSSINI, GREINER, AND MORDES ability to induce unresponsiveness. Transplant. Proc. 17: 393–396, 1985. BELLGRAU, D., D. GOLD, H. SELAWRY, J. MOORE, A. FRANZUSOFF, AND R. C. DUKE. A role for CD95 ligand in preventing graft rejection. Nature 377: 630–632, 1995. BENICHOU, G., AND E. V. FEDOSEYEVA. The contribution of peptides to T cell allorecognition and allograft rejection. Int. Rev. Immunol. 13: 231–243, 1996. BENICHOU, G., R. C. TAM, L. R. SOARES, AND E. V. FEDOSEYEVA. Indirect T-cell allorecognition: perspectives for peptide-based therapy in transplantation. Immunol. Today 18: 67–71, 1997. BEVAN, M. J. Antigen presentation to cytotoxic T lymphocytes in vivo. J. Exp. Med. 182: 639–641, 1995. BEVERLY, B., S. M. KANG, M. J. LENARDO, AND R. H. SCHWARTZ. Reversal of in vitro T cell clonal anergy by IL-2 stimulation. Int. Immunol. 4: 661–671, 1992. BILLINGHAM, R. E., L. BRENT, AND P. B. MEDAWAR. Acquired immunological tolerance. Nature 172: 603–604, 1953. BINDER, J., M. LEHMANN, E. GRASER, W. W. HANCOCK, B. WATSCHINGER, K. ONODERA, M. H. SAYEGH, H. D. VOLK, AND J. W. KUPIEC-WEGLINSKI. The effects of nondepleting CD4 targeted therapy in presensitized rat recipients of cardiac allografts. Transplantation 61: 804–811, 1996. BISHOP, G. A., G. W. MCCAUGHAN, J. SUN, AND A. G. SHEIL. Microchimerism and transplant tolerance. Immunol. Today 18: 455– 456, 1997. BISHOP, G. A., J. SUN, A. G. R. SHEIL, AND G. W. MCCAUGHAN. High dose/activation-associated tolerance: a mechanism for allograft tolerance. Transplantation 64: 1377–1382, 1997. BJÖRCK, P., J. BANCHEREAU, AND L. FLORES-ROMO. CD40 ligation counteracts Fas-induced apoptosis of human dendritic cells. Int. Immunol. 9: 365–372, 1997. BLACKMAN, M., J. KAPPLER, AND P. MARRACK. The role of the T cell receptor in positive and negative selection of developing T cells. Science 248: 1335–1341, 1990. BLAZAR, B. R., P. A. TAYLOR, A. PANOSKALTSIS-MORTARI, J. BUHLMAN, J. C. XU, R. A. FLAVELL, R. KORNGOLD, R. NOELLE, AND D. A. VALLERA. Blockade of CD40 ligand-CD40 interaction impairs CD4/ T cell-mediated alloreactivity by inhibiting mature donor T cell expansion and function after bone marrow transplantation. J. Immunol. 158: 29–39, 1997. BLUESTONE, J. A. New perspectives of CD28-B7-mediated T cell costimulation. Immunity 2: 555–559, 1995. BLUMBERG, N., AND J. M. HEAL. Transfusion-associated immunomodulation. In: Scientific Basis of Transfusion Medicine, edited by K. C. Anderson and P. M. Ness. Philadelphia, PA: Saunders, 1994, p. 580–598. BLUMBERG, N., AND J. M. HEAL. Immunomodulation by blood transfusion: an evolving scientific and clinical challenge. Am. J. Med. 101: 299–308, 1996. BLUMBERG, N., AND J. M. HEAL. The transfusion immunomodulation theory: the Th1/Th2 paradigm and an analogy with pregnancy as a unifying mechanism. Semin. Hematol. 33: 329–340, 1996. BOITARD, C., A. BENDELAC, M. F. RICHARD, C. CARNAUD, AND J. F. BACH. Prevention of diabetes in nonobese diabetic mice by anti-I-A monoclonal antibodies: transfer of protection by splenic T cells. Proc. Natl. Acad. Sci. USA 85: 9719–9723, 1988. BOLLING, S. F., H. LIN, R. Q. WEI, P. LINSLEY, AND L. A. TURKA. The effect of combination cyclosporine and CTLA4-Ig therapy on cardiac allograft survival. J. Surg. Res. 57: 60–64, 1994. BORRIELLO, F., G. J. FREEMAN, S. EDELHOFF, C. M. DISTECHE, L. M. NADLER, AND A. H. SHARPE. Characterization of the murine B7–1 genomic locus reveals an additional exon encoding an alternative cytoplasmic domain and a chromosomal location of chromosome 16, band B5. J. Immunol. 153: 5038–5048, 1994. BRADLEY, L. M., K. YOSHIMOTO, AND S. L. SWAIN. The cytokines IL-4, IFN-gamma, and IL-12 regulate the development of subsets of memory effector helper T cells in vitro. J. Immunol. 155: 1713– 1724, 1995. BRAUN, M. Y., A. MCCORMACK, G. WEBB, AND J. R. BATCHELOR. Mediation of acute but not chronic rejection of MHC-incompatible rat kidney grafts by alloreactive CD4 T cells activated by the direct pathway of sensitization. Transplantation 55: 177–182, 1993. / 9j0d$$ja01 P26-8 Volume 79 43. BRETSCHER, P., AND M. COHN. A theory of self-nonself discrimination. Science 169: 1042–1049, 1970. 44. BRUNET, J. F., F. DENIZOT, M. F. LUCIANI, M. ROUX-DOSSETO, M. SUZAN, M. G. MATTEI, AND P. GOLSTEIN. A new member of the immunoglobulin superfamily–CTLA-4. Nature 328: 267–270, 1987. 45. BUHLMANN, J. E., T. M. FOY, A. ARUFFO, K. M. CRASSI, J. A. LEDBETTER, W. R. GREEN, J. C. XU, L. D. SHULTZ, D. C. ROOPENIAN, R. A. FLAVELL, L. FAST, AND R. J. NOELLE. In the absence of a CD40 signal, B cells are tolerogenic. Immunity 2: 645–653, 1995. 46. BURKE, G. W., C. RICORDI, T. KARATZAS, M. CARRENO, M. MARKOU, R. CIROCCO, G. CIANCIO, T. QIAN, G. SELVAGGI, R. ALEJANDRO, J. S. SKYLER, AND D. ROTH. Donor bone marrow infusion in simultaneous pancreas/kidney transplantation with OKT3 induction: evidence for augmentation of chimerism. Transplant. Proc. 29: 1207–1208, 1997. 47. BURNET, F. M. The Integrity of the Body. A Discussion of Modern Immunological Ideas. Cambridge, MA: Harvard Univ. Press, 1962. 48. CALCINARO, F., K. J. LAFFERTY, AND N. N. SHEHADEH. Inflammatory mediators and development of autoimmune diabetes. In: Type I Diabetes. Molecular, Cellular, and Clinical Immunology, edited by G. S. Eisenbarth and K. J. Lafferty. Oxford, UK: Oxford Univ. Press, 1996, p. 91–117. 49. CAMPOS, L., E. J. ALFREY, A. M. POSSELT, J. S. ODORICO, C. F. BARKER, AND A. NAJI. Prolonged survival of rat orthotopic liver allografts after intrathymic inoculation of donor-strain cells. Transplantation 55: 866–870, 1993. 50. CAMPOS, L., A. M. POSSELT, B. C. DELI, G. L. MAYO, K. PETE, C. F. BARKER, AND A. NAJI. The failure of intrathymic transplantation of nonimmunogenic islet allografts to promote induction of donor-specific unresponsiveness. Transplantation 57: 950–953, 1994. 51. CARAYANNIOTIS, G., S. R. MASTERS, AND R. J. NOELLE. Suppression of murine thyroiditis via blockade of the CD40-CD40L interaction. Immunology 90: 421–426, 1997. 52. CARBONE, E., G. RUGGIERO, G. TERRAZZANO, C. PALOMBA, C. MANZO, S. FONTANA, H. SPITS, K. KÄRRE, AND S. ZAPPACOSTA. A new mechanism of NK cell cytotoxicity activation: the CD40-CD40 ligand interaction. J. Exp. Med. 185: 2053–2060, 1997. 53. CARROLL, P. B., P. FONTES, A. S. RAO, C. RICORDI, H. L. R. RILO, A. ZEEVI, M. TRUCCO, R. SHAPIRO, W. B. RYBKA, V. SCANTLEBURY, F. S. DODSON, AND A. G. TZAKIS. Simultaneous solid organ, bone marrow, and islet allotransplantation in type I diabetic patients. Transplant. Proc. 26: 3523–3524, 1994. 54. CHAHINE, A. A., M. YU, M. M. MCKERNAN, C. STOECKERT, AND H. T. LAU. Immunomodulation of pancreatic islet allografts in mice with CTLA4Ig secreting muscle cells. Transplantation 59: 1313– 1318, 1995. 55. CHAN, A. C., D. M. DESAI, AND A. WEISS. The role of protein tyrosine kinases and protein tyrosine phosphatases in T cell antigen receptor signal transduction. Annu. Rev. Immunol. 12: 555–592, 1994. 56. CHAN, S. Y., L. A. DEBRUYNE, R. E. GOODMAN, E. J. EICHWALD, / AND D. K. BISHOP. In vivo depletion of CD8 T cells results in Th2 cytokine production and alternate mechanisms of allograft rejection. Transplantation 59: 1155–1161, 1995. 57. CHANG, C. H., M. FURUE, AND K. TAMAKI. B7–1 expression of Langerhans cells is up-regulated by proinflammatory cytokines, and is down-regulated by interferon-gamma or by interleukin-10. Eur. J. Immunol. 25: 394–398, 1995. 58. CHARLTON, B., H. AUCHINCLOSS, JR., AND C. G. FATHMAN. Mechanisms of transplantation tolerance. Annu. Rev. Immunol. 12: 707–734, 1994. 59. CHEN, C., AND N. NABAVI. In vitro induction of T cell anergy by blocking B7 and early T cell costimulatory molecule ETC-1/B7–2. Immunity 1: 147–154, 1994. 60. CHEN, N., Q. GAO, AND E. H. FIELD. Expansion of memory Th2 cells over Th1 cells in neonatal primed mice. Transplantation 60: 1187–1193, 1995. 61. CHEN, Y., J. INOBE, R. MARKS, P. GONNELLA, V. K. KUCHROO, AND H. L. WEINER. Peripheral deletion of antigen-reactive T cells in oral tolerance. Nature 376: 177–180, 1995. 12-09-98 00:40:31 pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION 62. CHEN, Y., V. K. KUCHROO, J. INOBE, D. A. HAFLER, AND H. L. WEINER. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science 265: 1237– 1240, 1994. 63. CHEN, Y. H., J. INOBE, V. K. KUCHROO, J. L. BARON, C. A. JANEWAY, AND H. L. WEINER. Oral tolerance in myelin basic protein Tcell receptor transgenic mice: suppression of autoimmune encephalomyelitis and dose-dependent induction of regulatory cells. Proc. Natl. Acad. Sci. USA 93: 388–391, 1996. 64. CHERVONSKY, A. V., Y. WANG, F. S. WONG, I. VISINTIN, R. A. FLAVELL, C. A. JANEWAY, JR., AND L. A. MATIS. The role of Fas in autoimmune diabetes. Cell 89: 17–24, 1997. 65. CHOI, Y., J. W. KAPPLER, AND P. MARRACK. A superantigen encoded in the open reading frame of the 3* long terminal repeat of mouse mammary tumour virus. Nature 350: 203–207, 1991. 66. CLARK, L. B., T. M. FOY, AND R. J. NOELLE. CD40 and its ligand. Adv. Immunol. 63: 43–78, 1996. 67. COMMITTEE ON XENOGRAFT TRANSPLANTATION. Ethical issues and public policy. In: Xenotransplantation: Science, Ethics, and Public Policy. Washington, DC: National Academy Press, 1996, p. 1–126. 68. COULOMBE, M., H. YANG, S. GUERDER, R. A. FLAVELL, K. J. LAFFERTY, AND R. G. GILL. Tissue immunogenicity: the role of MHC antigen and the lymphocyte costimulator B7–1. J. Immunol. 157: 4790–4795, 1996. 69. CRAIU, A., T. AKOPIAN, A. GOLDBERG, AND K. L. ROCK. Two distinct proteolytic processes in the generation of a major histocompatibility complex class I-presented peptide. Proc. Natl. Acad. Sci. USA 94: 10850–10855, 1997. 70. CROFT, M., D. D. DUNCAN, AND S. L. SWAIN. Response of naive antigen-specific CD4/ T cells in vitro: characteristics and antigenpresenting cell requirements. J. Exp. Med. 176: 1431–1437, 1992. 71. CUTURI, M. C., R. JOSIEN, P. DOUILLARD, C. PANNETIER, D. CANTAROVICH, H. SMIT, S. MENORET, P. POULETTY, C. CLAYBERGER, AND J. P. SOULILLOU. Prolongation of allogeneic heart graft survival in rats by administration of a peptide (aa. 75–84) from the alpha 1 helix of the first domain of HLA-B7 01. Transplantation 59: 661–669, 1995. 72. DAIKH, D. I., B. K. FINCK, P. S. LINSLEY, D. HOLLENBAUGH, AND D. WOFSY. Long-term inhibition of murine lupus by brief simultaneous blockade of the B7/CD28 and CD40/gp39 costimulation pathways. J. Immunol. 159: 3104–3108, 1997. 73. DALLMAN, M. J., O. SHIHO, T. H. PAGE, K. J. WOOD, AND P. J. MORRIS. Peripheral tolerance to alloantigen results for altered regulation of the interleukin 2 pathway. J. Exp. Med. 173: 79–87, 1991. 74. DALLMAN, M. J., K. J. WOOD, AND P. J. MORRIS. Specific cytotoxic T cells are found in the nonrejected kidneys of blood-transfused rats. J. Exp. Med. 165: 566–571, 1987. 75. DANTAL, J., M. HOURMANT, D. CANTAROVICH, M. GIRAL, G. BLANCHO, B. DRENO, AND J. P. SOULILLOU. Effect of long term immunosuppression in kidney-graft recipients on cancer incidence: randomized comparison of two cyclosporin regimens. Lancet 351: 623–628, 1998. 76. DARBY, C. R., P. J. MORRIS, AND K. J. WOOD. Evidence that longterm cardiac allograft survival induced by anti-CD4 monoclonal antibody does not require depletion of CD4/ T cells. Transplantation 54: 483–490, 1992. 77. DATTA, S. K., AND S. L. KALLED. CD40-CD40 ligand interaction in autoimmune disease. Arthritis Rheum. 40: 1735–1745, 1997. 78. DAVIES, J. D., G. MARTIN, J. PHILLIPS, S. E. MARSHALL, S. P. COBBOLD, AND H. WALDMANN. T-cell regulation in adult transplantation tolerance. J. Immunol. 157: 529–533, 1996. 79. DEL PRETE, G., M. DE CARLI, F. ALMERIGOGNA, M. G. GIUDIZI, R. BIAGIOTTI, AND S. ROMAGNANI. Human IL-10 is produced by both type 1 helper (Th1) and type 2 helper (Th2) T cell clones and inhibits their antigen-specific proliferation and cytokine production. J. Immunol. 150: 353–360, 1993. 80. DEMETRIS, A. J., N. MURASE, Q. YE, F. H. GALVAO, C. RICHERT, R. SAAD, S. PHAM, R. J. DUQUESNOY, A. ZEEVI, J. J. FUNG, AND T. E. STARZL. Analysis of chronic rejection and obliterative arteriopathy. Possible contributions of donor antigen-presenting cells and lymphatic disruption. Am. J. Pathol. 150: 563–578, 1997. 81. DE WAAL MALEFYT, R., J. HAANEN, H. SPITS, M. G. RONCAR- / 9j0d$$ja01 P26-8 82. 83. 84. 85. 86. 87. 88. 89. 90. 91. 92. 93. 94. 95. 96. 97. 98. 99. 12-09-98 00:40:31 131 OLO, A. TE VELDE, C. FIGDOR, K. JOHNSON, R. KASTELEIN, H. YSSEL, AND J. E. DE VRIES. Interleukin 10 (IL-10) and viral IL10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J. Exp. Med. 174: 915–924, 1991. DE WAAL MALEFYT, R., H. YSSEL, AND J. E. DE VRIES. Direct effects of IL-10 on subsets of human CD4/ T cell clones and resting T cells. Specific inhibition of IL-2 production and proliferation. J. Immunol. 150: 4754–4765, 1993. DING, L., P. S. LINSLEY, L. Y. HUANG, R. N. GERMAIN, AND E. M. SHEVACH. IL-10 inhibits macrophage costimulatory activity by selectively inhibiting the up-regulation of B7 expression. J. Immunol. 151: 1224–1234, 1993. DONO, K., M. L. WOOD, H. OZATO, I. OTSU, R. GOTTSCHALK, T. MAKI, AND A. P. MONACO. Marked prolongation of rat skin xenografts induced by intrathymic injection of xenogeneic splenocytes and a short course of rapamycin in antilymphocyte serumtreated mice. Transplantation 59: 929–932, 1995. DORSCH, S., AND B. ROSER. Suppressor cells in transplantation tolerance. I. Analysis of the suppressor status of neonatally and adoptively tolerized rats. Transplantation 33: 518–524, 1982. DOUILLARD, P., C. PANNETIER, R. JOSIEN, S. MENORET, P. KOURILSKY, J. P. SOULILLOU, AND M. C. CUTURI. Donor-specific blood transfusion-induced tolerance in adult rats with a dominant TCR-Vbeta rearrangement in heart allografts. J. Immunol. 157: 1250–1260, 1996. DUBEY, C., M. CROFT, AND S. L. SWAIN. Costimulatory requirements of naive CD4/ T cells: ICAM-1 or B7–1 can costimulate naive CD4 T cell activation but both are required for optimum response. J. Immunol. 155: 45–57, 1995. DURIE, F. H., A. ARUFFO, J. LEDBETTER, K. M. CRASSI, W. R. GREEN, L. D. FAST, AND R. J. NOELLE. Antibody to the ligand of CD40, gp39, blocks the occurrence of the acute and chronic forms of graft-vs-host disease. J. Clin. Invest. 94: 1333–1338, 1994. DURIE, F. H., R. A. FAVA, T. M. FOY, A. ARUFFO, J. A. LEDBETTER, AND R. J. NOELLE. Prevention of collagen-induced arthritis with an antibody to gp39, the ligand for CD40. Science 261: 1328– 1330, 1993. EARLY, G. S., W. G. ZHAO, AND C. M. BURNS. Anti-CD40 ligand antibody treatment prevents the development of lupus-like nephritis in a subset of New Zealand black 1 New Zealand white mice: response correlates with the absence of an anti-antibody response. J. Immunol. 157: 3159–3164, 1996. EYNON, E. E., AND D. C. PARKER. Small B cells as antigen-presenting cells in the induction of tolerance to soluble protein antigens. J. Exp. Med. 175: 131–138, 1992. EYNON, E. E., AND D. C. PARKER. Parameters of tolerance induction by antigen targeted to B lymphocytes. J. Immunol. 151: 2958– 2964, 1993. FAUSTMAN, D. Strategies for circumventing transplant rejection: modification of cells, tissues and organs. Trends Biotechnol. 13: 100–105, 1995. FAUSTMAN, D., AND C. COE. Prevention of xenograft rejection by masking donor HLA class I antigens. Science 252: 1700–1702, 1991. FAUSTMAN, D., V. HAUPTFELD, P. LACY, AND J. DAVIE. Prolongation of murine islet allograft survival by pretreatment of islets with antibody directed to Ia determinants. Proc. Natl. Acad. Sci. USA 78: 5156–5159, 1981. FECHNER, J. H., JR., D. J. VARGO, E. K. GEISSLER, C. GRAEB, J. WANG, M. J. HANAWAY, D. I. WATKINS, M. PIEKARCZYK, D. M. NEVILLE, JR., AND S. J. KNECHTLE. Split tolerance induced by immunotoxin in a rhesus kidney allograft model. Transplantation 63: 1339–1345, 1997. FIELD, E. H., Q. GAO, N. X. CHEN, AND T. M. ROUSE. Balancing the immune system for tolerance: a case for regulatory CD4 cells. Transplantation 64: 1–7, 1997. FINKELMAN, F. D., A. LEES, AND S. C. MORRIS. Antigen presentation by B lymphocytes to CD4/ T lymphocytes in vivo: importance for B lymphocyte and T lymphocyte activation. Semin. Immunol. 4: 247–255, 1992. FIORETTO, P., M. W. STEFFES, D. E. R. SUTHERLAND, F. C. GOETZ, AND M. MAUER. Reversal of lesions of diabetic nephropa- pra APS-Phys Rev 132 100. 101. 102. 103. 104. 105. 106. 107. 108. 109. 110. 111. 112. 113. 114. 115. 116. 117. 118. 119. ROSSINI, GREINER, AND MORDES thy after pancreas transplantation. N. Engl. J. Med. 339: 69–75, 1998. FISHMAN-LOBELL, J., A. FRIEDMAN, AND H. L. WEINER. Different kinetic patterns of cytokine gene expression in vivo in orally tolerant mice. Eur. J. Immunol. 24: 2720–2724, 1994. FONTES, P., A. S. RAO, A. J. DEMETRIS, A. ZEEVI, M. TRUCCO, P. CARROLL, W. RYBKA, W. A. RUDERT, C. RICORDI, F. DODSON, R. SHAPIRO, AND A. TZAKIS. Bone marrow augmentation of donorcell chimerism in kidney, liver, heart, and pancreas islet transplantation. Lancet 344: 151–155, 1994. FORSTHUBER, T., H. C. YIP, AND P. V. LEHMANN. Induction of TH1 and TH2 immunity in neonatal mice. Science 271: 1728–1730, 1996. FOY, T. M., A. ARUFFO, J. BAJORATH, J. E. BUHLMANN, AND R. J. NOELLE. Immune regulation by CD40 and its ligand gp39. Annu. Rev. Immunol. 14: 591–617, 1996. FOY, T. M., M. MCILRAITH, S. R. MASTERS, J. J. DUNN, A. A. ROSSINI, L. D. SHULTZ, R. A. HESSELTON, E. J. WAGAR, P. E. LIPSKY, R. J. NOELLE, AND D. L. GREINER. Blockade of CD40-CD154 interferes with human T cell engraftment in scid mice. Cell Transplant. 7: 25–35, 1998. FRASCA, L., P. CARMICHAEL, R. LECHLER, AND G. LOMBARDI. Anergic T cells effect linked suppression. Eur. J. Immunol. 27: 3191–3197, 1997. FREEMAN, G. J., F. BORRIELLO, R. J. HODES, H. REISER, K. S. HATHCOCK, G. LASZLO, A. J. MCKNIGHT, J. KIM, L. DU, D. B. LOMBARD, G. S. GRAY, AND L. M. NADLER. Uncovering of functional alternative CTLA-4 counter-receptor in B7-deficient mice. Science 262: 907–909, 1993. FREEMAN, G. J., V. A. BOUSSIOTIS, A. ANUMANTHAN, G. M. BERNSTEIN, X. Y. KE, P. D. RENNERT, G. S. GRAY, J. G. GRIBBEN, AND L. M. NADLER. B7–1 and B7–2 do not deliver identical costimulatory signals, since B7–2 but not B7–1 preferentially costimulates the initial production of IL-4. Immunity 2: 523–532, 1995. FUCHS, E. J., AND P. MATZINGER. B cells turn off virgin but not memory T cells. Science 258: 1156–1159, 1992. GALILI, U. Interaction of the natural anti-Gal antibody with a-galactosyl epitopes: a major obstacle for xenotransplantation in humans. Immunol. Today 14: 480–482, 1993. GALILI, U., AND K. L. MATTA. Inhibition of anti-Gal IgG binding to porcine endothelial cells by synthetic oligosaccharides. Transplantation 62: 256–262, 1996. GAO, Q., N. CHEN, T. M. ROUSE, AND E. H. FIELD. The role of interleukin-4 in the induction phase of allogeneic neonatal tolerance. Transplantation 62: 1847–1854, 1996. GARSIDE, P., E. INGULLI, R. R. MERICA, J. G. JOHNSON, R. J. NOELLE, AND M. K. JENKINS. Visualization of specific B and T lymphocyte interactions in the lymph node. Science 281: 96–99, 1998. GARSIDE, P., M. STEEL, E. A. WORTHEY, A. SATOSKAR, J. ALEXANDER, H. BLUETHMANN, F. Y. LIEW, AND A. M. MOWAT. T helper 2 cells are subject to high dose oral tolerance and are not essential for its induction. J. Immunol. 154: 5649–5655, 1995. GERRITSE, K., J. D. LAMAN, R. J. NOELLE, A. ARUFFO, J. A. LEDBETTER, W. J. A. BOERSMA, AND E. CLAASSEN. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. USA 93: 2499–2504, 1996. GERY, I., AND J. W. STREILEIN. Autoimmunity in the eye and its regulation. Curr. Opin. Immunol. 6: 938–945, 1994. GILBERT, K. M., AND W. O. WEIGLE. Tolerogenicity of resting and activated B cells. J. Exp. Med. 179: 249–258, 1994. GILL, R. G. The immunobiology of pancreatic islet transplantation. In: Type I Diabetes. Molecular, Cellular, and Clinical Immunology, edited by G. S. Eisenbarth and K. J. Lafferty. Oxford, UK: Oxford Univ. Press, 1996, p. 118–133. GORDON, E. J., T. G. MARKEES, N. E. PHILLIPS, R. J. NOELLE, L. D. SHULTZ, J. P. MORDES, A. A. ROSSINI, AND D. L. GREINER. Prolonged survival of rat islet and skin xenografts in mice treated with donor splenocytes and anti-CD154 monoclonal antibody. Diabetes 47: 1199–1206, 1998. GOSS, J. A., M. W. FLYE, AND P. E. LACY. Successful transfer of immune unresponsiveness to concordant rat islet xenografts. Transplantation 61: 9–13, 1996. / 9j0d$$ja01 P26-8 Volume 79 120. GOSS, J. A., Y. NAKAFUSA, E. H. FINKE, M. W. FLYE, AND P. E. LACY. Induction of tolerance to islet xenografts in a concordant rat-to-mouse model. Diabetes 43: 16–23, 1994. 121. GOSS, J. A., Y. NAKAFUSA, AND M. W. FLYE. Intrathymic injection of donor alloantigens induces donor-specific vascularized allograft tolerance without immunosuppression. Ann. Surg. 216: 409–416, 1992. 122. GREWAL, I. S., AND R. A. FLAVELL. The CD40 ligand: at the center of the immune universe. Immunol. Res. 16: 59–70, 1997. 123. GREWAL, I. S., H. G. FOELLMER, K. D. GREWAL, J. C. XU, F. HARDARDOTTIR, J. L. BARON, C. A. JANEWAY, JR., AND R. A. FLAVELL. Requirement for CD40 ligand in costimulation induction, T cell activation, and experimental allergic encephalomyelitis. Science 273: 1864–1867, 1996. 124. GREWAL, I. S., J. C. XU, AND R. A. FLAVELL. Impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature 378: 617–620, 1995. 125. GRIBBEN, J. G., G. J. FREEMAN, V. A. BOUSSIOTIS, P. RENNERT, C. L. JELLIS, E. GREENFIELD, M. BARBER, V. A. RESTIVO, JR., X. KE, G. S. GRAY, AND L. M. NADLER. CTLA4 mediates antigenspecific apoptosis of human T cells. Proc. Natl. Acad. Sci. USA 92: 811–815, 1995. 126. GRIFFITH, T. S., T. BRUNNER, S. M. FLETCHER, D. R. GREEN, AND T. A. FERGUSON. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science 270: 1189–1192, 1995. 127. GRIFFITH, T. S., AND T. A. FERGUSON. The role of FasL-induced apoptosis in immune privilege. Immunol. Today 18: 240–244, 1997. 128. GROTH, C. G., O. KORSGREN, A. TIBELL, J. TOLLEMAR, E. MOLLER, J. BOLINDER, J. OSTMAN, F. P. REINHOLT, C. HELLERSTROM, AND A. ANDERSSON. Transplantation of porcine fetal pancreas to diabetic patients. Lancet 344: 1402–1404, 1994. 129. GROUX, H., M. BIGLER, J. E. DE VRIES, AND M. G. RONCAROLO. Interleukin-10 induces a long-term antigen-specific anergic state in human CD4/ T cells. J. Exp. Med. 184: 19–29, 1996. 130. GRUSBY, M. J., H. AUCHINCLOSS, JR., R. LEE, R. S. JOHNSON, J. P. SPENCER, M. ZIJLSTRA, R. JAENISCH, V. E. PAPAIOANNOU, AND L. H. GLIMCHER. Mice lacking major histocompatibility complex class I and class II molecules. Proc. Natl. Acad. Sci. USA 90: 3913–3917, 1993. 131. GUERDER, S., AND P. MATZINGER. A fail-safe mechanism for maintaining self-tolerance. J. Exp. Med. 176: 553–564, 1992. 132. GUERETTE, B., D. SKUK, F. CELESTIN, C. HUARD, F. TARDIF, I. ASSELIN, B. ROY, M. GOULET, R. ROY, M. ENTMAN, AND J. P. TREMBLAY. Prevention by anti-LFA-1 of acute myoblast death following transplantation. J. Immunol. 159: 2522–2531, 1997. 133. GUO, Y., Y. WU, M. ZHAO, X. P. KONG, AND Y. LIU. Mutational analysis and an alternatively spliced product of B7 defines its CD28/ CTLA4-binding site on immunoglobulin C-like domain. J. Exp. Med. 181: 1345–1355, 1995. 134. GUTTMANN, R. D. Developing ways of reducing allograft immunogenicity. CMA J. 124: 143–145, 1981. 135. GUTTMANN, R. D., AND R. D. C. FORBES. Passenger leukocytes revisited. Transplant. Proc. 16: 37–39, 1984. 136. HALE, D. A., R. GOTTSCHALK, T. MAKI, AND A. P. MONACO. Use of CTLA4-Ig in combination with conventional immunosuppressive agents to prolong allograft survival. Transplantation 64: 897–900, 1997. 137. HALL, B. M., M. E. JELBART, K. E. GURLEY, AND S. E. DORSCH. Specific unresponsiveness in rats with prolonged cardiac allograft survival after treatment with cyclosporine. Mediation of specific suppression by T helper/inducer cells. J. Exp. Med. 162: 1683–1694, 1985. 138. HALLORAN, P. F., AND S. L. LUI. Approved immunosuppressants. In: Primer on Transplantation, edited by D. J. Norman and W. N. Suki. Thorofare, NJ: Am. Soc. Transplant Physicians, 1998, p. 93– 102. 139. HANCOCK, G. E., Z. A. COHN, AND G. KAPLAN. The generation of antigen-specific, major histocompatibility complex-restricted cytotoxic T lymphocytes of the CD4/ phenotype. Enhancement by the cutaneous administration of interleukin 2. J. Exp. Med. 169: 909– 919, 1989. 140. HARGREAVES, R. G., N. J. BORTHWICK, M. S. G. MONTANI, E. PICCOLELLA, P. CARMICHAEL, R. I. LECHLER, A. N. AKBAR, AND 12-09-98 00:40:31 pra APS-Phys Rev January 1999 141. 142. 143. 144. 145. 146. 147. 148. 149. 150. 151. 152. 153. 154. 155. 156. 157. 158. 159. INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION G. LOMBARDI. Dissociation of T cell anergy from apoptosis by blockade of Fas/Apo-1 (CD95) signaling. J. Immunol. 158: 3099– 3107, 1997. HARP, J. A., AND S. J. EWALD. Modulation of in vitro immune responses by monoclonal antibody to T200 antigen. Cell. Immunol. 81: 71–80, 1983. HART, D. N. J. Dendritic cells: unique leukocyte populations which control the primary immune response. Blood 90: 3245–3287, 1997. HATHCOCK, K. S., G. LASZLO, H. B. DICKLER, J. BRADSHAW, P. LINSLEY, AND R. J. HODES. Identification of an alternative CTLA4 ligand costimulatory for T cell activation. Science 262: 905–907, 1993. HATHCOCK, K. S., G. LASZLO, C. PUCILLO, P. LINSLEY, AND R. J. HODES. Comparative analysis of B7–1 and B7–2 costimulatory ligands: expression and function. J. Exp. Med. 180: 631–640, 1994. HAYRY, P. Pathophysiology of chronic rejection. Transplant. Proc. 28: 7–10, 1996. HAYRY, P., H. ISONIEMI, S. YILMAZ, A. MENNANDER, K. LEMSTROM, A. RAISANEN-SOKOLOWSKI, P. KOSKINEN, J. USTINOV, I. LAUTENSCHLAGER, AND E. TASKINEN. Chronic allograft rejection. Immunol. Rev. 134: 33–81, 1993. HEATH, W. R., J. ALLISON, M. W. HOFFMANN, G. SCHÖNRICH, G. HÄMMERLING, B. ARNOLD, AND J. F. A. P. MILLER. Autoimmune diabetes as a consequence of locally produced interleukin-2. Nature 359: 547–549, 1992. HENN, V., J. R. SLUPSKY, M. GRÄFE, I. ANAGNOSTOPOULOS, R. FÖRSTER, G. MÜLLER-BERGHAUS, AND R. A. KROCZEK. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 391: 591–594, 1998. HERING, B. J. Islet xenotransplantation. In: Pancreatic Islet Cell Transplantation: 1892–1992, One Century of Transplantation for Diabetes, edited by C. Ricordi. Austin, TX: Landes, 1992, p. 313– 335. HOFFMANN, M. W., J. ALLISON, AND J. F. A. P. MILLER. Tolerance induction by thymic medullary epithelium. Proc. Natl. Acad. Sci. USA 89: 2526–2530, 1992. HORI, S., S. SATO, S. KITAGAWA, T. AZUMA, S. KOKUDO, T. HAMAOKA, AND H. FUJIWARA. Tolerance induction of allo-class H-2 antigen-reactive L3T4/ helper T cells and prolonged survival of the corresponding class II H-2-disparate skin graft. J. Immunol. 143: 1447–1452, 1989. IDO, Y., A. VINDIGNI, K. CHANG, L. STRAMM, R. CHANCE, W. F. HEATH, R. D. DIMARCHI, E. DI CERA, AND J. R. WILLIAMSON. Prevention of vascular and neural dysfunction in diabetic rats by C-peptide. Science 277: 563–566, 1997. ILDSTAD, S. T., D. M. WREN, S. S. BOGGS, M. L. HRONAKES, F. VECCHINI, AND M. R. M. VAN DEN BRINK. Cross-species bone marrow transplantation: evidence for tolerance induction, stem cell engraftment, and maturation of T lymphocytes in a xenogeneic stromal environment (rat to mouse). J. Exp. Med. 174: 467–478, 1991. INOBE, M., N. AOKI, P. S. LINSLEY, J. A. LEDBETTER, R. ABE, M. MURAKAMI, AND T. UEDE. The role of the B7–1a molecule, an alternatively spliced form of murine B7–1 (CD80), on T cell activation. J. Immunol. 157: 582–588, 1996. INOBE, M., P. S. LINSLEY, J. A. LEDBETTER, Y. NAGAI, M. TAMAKOSHI, AND T. UEDE. Identification of an alternatively spliced form of the murine homologue of B7. Biochem. Biophys. Res. Commun. 200: 443–449, 1994. ISOBE, M., H. YAGITA, K. OKUMURA, AND A. IHARA. Specific acceptance of cardiac allograft after treatment with antibodies to ICAM-1 and LFA-1. Science 255: 1125–1127, 1992. IWATA, T., Y. KAMEI, S. ESAKI, T. TAKADA, S. TORII, A. YAMASHITA, S. TOMIDA, T. TAMATANI, M. MIYASAKA, AND Y. YOSHIKAI. Immunosuppression by anti-ICAM-1 and anti-LFA-1 monoclonal antibodies of free and vascularized skin allograft rejection. Immunobiology 195: 160–171, 1996. JENKINS, M. K., E. BURRELL, AND J. D. ASHWELL. Antigen presentation by resting B cells. Effectiveness at inducing T cell proliferation is determined by costimulatory signals, not T cell receptor occupancy. J. Immunol. 144: 1585–1590, 1990. JENKINS, M. K., AND R. H. SCHWARTZ. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell un- / 9j0d$$ja01 P26-8 133 responsiveness in vitro and in vivo. J. Exp. Med. 165: 302–319, 1987. 160. JONES, N. D., N. C. FLUCK, D. L. ROELEN, A. L. MELLOR, P. J. MORRIS, AND K. J. WOOD. Deletion of alloantigen-reactive thymocytes as a mechanism of adult tolerance induction following intrathymic antigen administration. Eur. J. Immunol. 27: 1591–1600, 1997. 161. JOSIEN, R., C. PANNETIER, P. DOUILLARD, D. CANTAROVICH, S. MENORET, L. BUGEON, P. KOURILSKY, J. P. SOULILLOU, AND M. C. CUTURI. Graft-infiltrating T helper cells, CD45RC phenotype, and Th1/Th2-related cytokines in donor-specific transfusion-induced tolerance in adult rats. Transplantation 60: 1131–1139, 1995. 162. KAGI, D., B. LEDERMANN, K. BURKI, P. SEILER, B. ODERMATT, K. J. OLSEN, PODACK, ER, R. M. ZINKERNAGEL, AND H. HENGARTNER. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 369: 31–37, 1994. 163. KANG, S. M., D. B. SCHNEIDER, Z. H. LIN, D. HANAHAN, D. A. DICHEK, P. G. STOCK, AND S. BAEKKESKOV. Fas ligand expression in islets of Langerhans does not confer immune privilege and instead targets them for rapid destruction. Nature Med. 3: 738– 743, 1997. 164. KAPPLER, J. W., N. ROEHM, AND P. MARRACK. T cell tolerance by clonal elimination in the thymus. Cell 49: 273–280, 1987. 165. KARANDIKAR, N. J., C. L. VANDERLUGT, T. L. WALUNAS, S. D. MILLER, AND J. A. BLUESTONE. CTLA-4: a negative regulator of autoimmune disease. J. Exp. Med. 184: 783–788, 1996. 166. KARMANN, K., W. MIN, W. C. FANSLOW, AND J. S. POBER. Activation and homologous desensitization of human endothelial cells by CD40 ligand, tumor necrosis factor, and interleukin 1. J. Exp. Med. 184: 173–182, 1996. 167. KAST, W. M., E. VAN TWUYVER, R. J. MOOIJAART, M. VERVELD, A. G. KAMPHUIS, C. J. MELIEF, AND L. P. DE WAAL. Mechanism of skin allograft enhancement across an H-2 class I mutant difference. Evidence for involvement of veto cells. Eur. J. Immunol. 18: 2105– 2108, 1988. 167a.KELSO, A. The enigma of cytokine redundancy. Immunol. Cell. Biol. 72: 97–101, 1994. 168. KENNEDY, M. K., K. M. MOHLER, K. D. SHANEBECK, P. R. BAUM, K. S. PICHA, C. A. OTTEN-EVANS, C. A. JANEWAY, JR., AND K. H. GRABSTEIN. Induction of B cell costimulatory function by recombinant murine CD40 ligand. Eur. J. Immunol. 24: 116–123, 1994. 169. KENYON, N., L. FERNANDEZ, M. MASETTI, R. LEHMANN, A. RANUNCOLI, M. CHATZIPETROU, D. HAN, M. OLIVEIRA, A. KIRK, D. HARLAN, L. INVERARDI, AND J. WAGNER. Humanized antiCD154 enhances islet allograft survival in non-human primates. Transplant. Proc. In press. 170. KERMAN, R. H., C. G. OROSZ, AND M. I. LORBER. Clinical relevance of anti-HLA antibodies pre and post transplant. Am. J. Med. Sci. 313: 275–278, 1997. 170a.KIMIKAWA, M., D. H. SACHS, R. B. COLVIN, A. BARTHOLOMEW, T. KAWAI, AND A. B. COSIMI. Modifications of the conditioning regimen for achieving mixed chimerism and donor-specific tolerance in cynomologus monkeys. Transplant 64: 709–716, 1997. 171. KIRK, A. D., D. M. HARLAN, N. N. ARMSTRONG, T. A. DAVIS, Y. C. DONG, G. S. GRAY, X. N. HONG, D. THOMAS, J. H. FECHNER, JR., AND S. J. KNECHTLE. CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc. Natl. Acad. Sci. USA 94: 8789– 8794, 1997. 172. KISIELOW, P., H. BLUTHMANN, U. D. STAERZ, M. STEINMETZ, AND H. VON BOEHMER. Tolerance in T-cell-receptor transgenic mice involves deletion of nonmature CD4/8/ thymocytes. Nature 333: 742–746, 1988. 173. KLEIN, J. Natural History of the Major Histocompatibility Complex. New York: Wiley, 1986, p. 1–775. 174. KLEIN, J. Immunology. New York: Blackwell Scientific, 1990, p. 460–476. 175. KNECHTLE, S. J., D. VARGO, J. FECHNER, Y. ZHAI, J. WANG, M. J. HANAWAY, J. SCHARFF, H. HU, L. KNAPP, D. WATKINS, AND D. M. NEVILLE, JR. FN18-CRM9 immunotoxin promotes tolerance in primate renal allografts. Transplantation 63: 1–6, 1997. 176. KNECHTLE, S. J., J. WANG, C. GRAEB, Y. ZHAI, X. HONG, J. H. FECHNER, JR., AND E. K. GEISSLER. Direct MHC class I comple- 12-09-98 00:40:31 pra APS-Phys Rev 134 177. 178. 179. 180. 181. 182. 183. 184. 185. 186. 187. 188. 189. 190. 191. 192. 193. 194. 195. ROSSINI, GREINER, AND MORDES mentary DNA transfer to thymus induces donor-specific unresponsiveness, which involves multiple immunologic mechanisms. J. Immunol. 159: 152–158, 1997. KOBATA, T., H. IKEDA, Y. OHNISHI, N. URUSHIBARA, T. A. TAKAHASHI, AND S. SEKIGUCHI. UV irradiation can induce in vitro clonal anergy in alloreactive cytotoxic T lymphocytes. Blood 82: 176–181, 1993. KORDOWER, J. H., T. B. FREEMAN, B. J. SNOW, F. J. VINGERHOETS, E. J. MUFSON, P. R. SANBERG, R. A. HAUSER, D. A. SMITH, G. M. NAUERT, AND D. P. PERL. Neuropathological evidence of graft survival and striatal reinnervation after the transplantation of fetal mesencephalic tissue in a patient with Parkinson’s disease. N. Engl. J. Med. 332: 1118–1124, 1995. KORDOWER, J. H., J. M. ROSENSTEIN, T. J. COLLIER, M. A. BURKE, E. Y. CHEN, J. M. LI, L. MARTEL, A. E. LEVEY, E. J. MUFSON, T. B. FREEMAN, AND C. W. OLANOW. Functional fetal nigral grafts in a patient with Parkinson’s disease: chemoanatomic, ultrastructural, and metabolic studies. J. Comp. Neurol. 370: 203–230, 1996. KOULMANDA, M., AND T. E. MANDEL. Effect of anti-CD4 and antiICAM MAb on survival of fetal pig pancreas grafts in NOD mice. Transplant. Proc. 26: 3466–3466, 1994. KOVACSOVICS-BANKOWSKI, M., AND K. L. ROCK. A phagosometo-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science 267: 243–246, 1995. KRIEGER, N. R., D. P. YIN, AND C. G. FATHMAN. CD4/ but not CD8/ cells are essential for allorejection. J. Exp. Med. 184: 2013– 2018, 1996. KRUISBEEK, A. M., AND D. AMSEN. Mechanisms underlying T-cell tolerance. Curr. Opin. Immunol. 8: 233–244, 1996. KRUMMEL, M. F., AND J. P. ALLISON. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 182: 459–465, 1995. KUCHROO, V. K., M. P. DAS, J. A. BROWN, A. M. RANGER, S. S. ZAMVIL, R. A. SOBEL, H. L. WEINER, N. NABAVI, AND L. H. GLIMCHER. B7–1 and B7–2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell 80: 707–718, 1995. KUPIEC-WEGLINSKI, J. W., B. WASOWSKA, I. PAPP, G. SCHMIDBAUER, M. H. SAYEGH, W. M. BALDWIN III, K. J. WIEDER, AND W. W. HANCOCK. CD4 mAb therapy modulates alloantibody production and intracardiac graft deposition in association with selective inhibition of Th1 lymphokines. J. Immunol. 151: 5053–5061, 1993. KWEON, M.-N., K. FUJIHASHI, J. L. VANCOTT, K. HIGUCHI, M. YAMAMOTO, J. R. MCGHEE, AND H. KIYONO. Lack of orally induced systemic unresponsiveness in IFN-gamma knockout mice. J. Immunol. 160: 1687–1693, 1998. LACY, P. E. Islet transplantation: the future. In: Pancreatic Islet Cell Transplantation: 1892–1992, One Century of Transplantation for Diabetes, edited by C. Ricordi.. Austin, TX: Landes, 1992, p. 394– 399. LACY, P. E. Status of islet cell transplantation. Diabetes Rev. 1: 76– 92, 1993. LACY, P. E., J. M. DAVIE, AND E. H. FINKE. Prolongation of islet allograft survival following in vitro culture (247) and a single injection of ALS. Science 204: 312–313, 1979. LAFFERTY, K. J. Islet cell transplantation as a therapy for type I diabetes mellitus. Diabetes Nutr. Metab. 2: 323–332, 1989. LAFFERTY, K. J., M. A. COOLEY, J. WOOLNOUGH, AND K. Z. WALKER. Thyroid allograft immunogenicity is reduced after a period in organ culture. Science 188: 259–261, 1975. LAFFERTY, K. J., S. J. PROWSE, C. J. SIMEONOVIC, AND H. S. WARREN. Immunobiology of tissue transplantation: a return to the passenger leukocyte concept. Annu. Rev. Immunol. 1: 143–173, 1983. LAKKIS, F. G., B. T. KONIECZNY, S. SALEEM, F. K. BADDOURA, P. S. LINSLEY, D. Z. ALEXANDER, R. P. LOWRY, T. C. PEARSON, AND C. P. LARSEN. Blocking the CD28-B7 T cell costimulation pathway induces long term cardiac allograft acceptance in the absence of IL-4. J. Immunol. 158: 2443–2448, 1997. LAMAN, J. D., E. CLAASSEN, AND R. J. NOELLE. Functions of CD40 and its ligand, gp39 (CD40L). Crit. Rev. Immunol. 16: 59–108, 1996. / 9j0d$$ja01 P26-8 Volume 79 196. LAMB, J. R., B. J. SKIDMORE, N. GREEN, J. M. CHILLER, AND M. FELDMANN. Induction of tolerance in influenza virus-immune T lymphocyte clones with synthetic peptides of influenza hemagglutinin. J. Exp. Med. 157: 1434–1447, 1983. 197. LANE, P., A. TRAUNECKER, S. HUBELE, S. INUI, A. LANZAVECCHIA, AND D. GRAY. Activated human T cells express a ligand for the human B cell-associated antigen CD40 which participates in T cell-dependent activation of B lymphocytes. Eur. J. Immunol. 22: 2573–2578, 1992. 198. LANOUE, A., C. BONA, H. VON BOEHMER, AND A. SARUKHAN. Conditions that induce tolerance in mature CD4/ T cells. J. Exp. Med. 185: 405–414, 1997. 199. LANZA, R. P., AND W. L. CHICK. Transplantation of encapsulated cells and tissues. Surgery 121: 1–9, 1997. 200. LARSEN, C. P., D. Z. ALEXANDER, D. HOLLENBAUGH, E. T. ELWOOD, S. C. RITCHIE, A. ARUFFO, R. HENDRIX, AND T. C. PEARSON. CD40-gp39 interactions play a critical role during allograft rejection. Suppression of allograft rejection by blockade of the CD40-gp39 pathway. Transplantation 61: 4–9, 1996. 201. LARSEN, C. P., E. T. ELWOOD, D. Z. ALEXANDER, S. C. RITCHIE, R. HENDRIX, C. TUCKER-BURDEN, H. R. CHO, A. ARUFFO, D. HOLLENBAUGH, P. S. LINSLEY, K. J. WINN, AND T. C. PEARSON. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature 381: 434–438, 1996. 202. LARSEN, C. P., S. C. RITCHIE, R. HENDRIX, P. S. LINSLEY, K. S. HATHCOCK, R. J. HODES, R. P. LOWRY, AND T. C. PEARSON. Regulation of immunostimulatory function and costimulatory molecule (B7–1 and B7–2) expression on murine dendritic cells. J. Immunol. 152: 5208–5219, 1994. 203. LAU, H., K. REEMTSMA, AND M. A. HARDY. Pancreatic islet allograft prolongation by donor-specific blood transfusions treated with ultraviolet irradiation. Science 221: 754–756, 1983. 204. LAU, H. T., M. YU, A. FONTANA, AND C. J. STOECKERT, JR. Prevention of islet allograft rejection with engineered myoblasts expressing FasL in mice. Science 273: 109–112, 1996. 205. LAZAROVITS, A. I., S. POPPEMA, M. J. WHITE, AND J. KARSH. Inhibition of alloreactivity in vitro by monoclonal antibodies directed against restricted isoforms of the leukocyte-common antigen (CD45). Transplantation 54: 724–729, 1992. 206. LAZAROVITS, A. I., S. POPPEMA, Z. ZHANG, M. KHANDAKER, C. E. LE FEUVRE, S. K. SINGHAL, B. M. GARCIA, N. OGASA, A. M. JEVNIKAR, M. J. WHITE, G. SINGH, AND C. R. STILLER. Prevention and reversal of renal allograft rejection by antibody against CD45RB. Nature 380: 717–720, 1996. 207. LECHLER, R. I., AND J. R. BATCHELOR. Restoration of immunogenicity to passenger cell-depleted kidney allograft by the addition of donor strain dendritic cells. J. Exp. Med. 155: 31–41, 1982. 208. LEE, R. S., M. J. GRUSBY, L. H. GLIMCHER, H. J. WINN, AND H. AUCHINCLOSS, JR. Indirect recognition by helper cells can induce donor-specific cytotoxic T lymphocytes in vivo. J. Exp. Med. 179: 865–872, 1994. 209. LEFKOWITH, J., G. SCHREINER, J. CORMIER, E. S. HANDLER, H. K. DRISCOLL, D. GREINER, J. P. MORDES, AND A. A. ROSSINI. Prevention of diabetes in the BB rat by essential fatty acid deficiency. Relationship between physiological and biochemical changes. J. Exp. Med. 171: 729–743, 1990. 210. LEFKOWITH, J. B., AND G. SCHREINER. Essential fatty acid deficiency depletes rat glomeruli of resident macrophages and inhibits angiotensin II-induced eicosanoid synthesis. J. Clin. Invest. 80: 947–956, 1987. 211. LEHMANN, M., E. GRASER, K. RISCH, W. W. HANCOCK, A. MULLER, B. KUTTLER, H. J. HAHN, J. W. KUPIEC-WEGLINSKI, J. W. BROCK, AND H. D. VOLK. Anti-CD4 monoclonal antibody-induced allograft tolerance in rats despite persistence of donor-reactive T cells. Transplantation 64: 1181–1187, 1997. 212. LEHMANN, R., N. KENYON, L. FERNANDEZ, M. MASETTI, M. OLIVEIRA-GANDIA, A. RANUNCOLI, G. IARIA, L. BURKLY, J. WAGNER, R. ALEJANDRO, AND C. RICORDI. Prevention of rejection allows for maintenance of islet mass at pre-pancreatectomy levels in Baboon (Papio hamadryas) recipients of islet allografts (Abstract). Diabetes 47, Suppl. 1: A75, 1998. 213. LENSCHOW, D. J., S. C. HO, H. SATTAR, L. RHEE, G. GRAY, N. NABAVI, K. C. HEROLD, AND J. A. BLUESTONE. Differential ef- 12-09-98 00:40:31 pra APS-Phys Rev January 1999 214. 215. 216. 217. 218. 219. 220. 221. 222. 223. 224. 225. 226. 227. 228. 229. 230. 231. INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION fects of anti-B7–1 and anti-B7–2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J. Exp. Med. 181: 1145–1155, 1995. LENSCHOW, D. J., Y. ZENG, J. R. THISTLETHWAITE, A. MONTAG, W. BRADY, M. G. GIBSON, P. S. LINSLEY, AND J. A. BLUESTONE. Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4Ig. Science 257: 789–792, 1992. LEVISETTI, M. G., P. A. PADRID, G. L. SZOT, N. MITTAL, S. M. MEEHAN, C. L. WARDRIP, G. S. GRAY, D. S. BRUCE, R. J. THISTLETHWAITE, JR., AND J. A. BLUESTONE. Immunosuppressive effects of human CTLA4Ig in a non-human primate model of allogeneic pancreatic islet transplantation. J. Immunol. 159: 5187–5191, 1997. LI, H., Y. L. COLSON, AND S. T. ILDSTAD. Mixed allogeneic chimerism achieved by lethal and nonlethal conditioning approaches induces donor-specific tolerance to simultaneous islet allografts. Transplantation 60: 523–529, 1995. LIBLAU, R. S., R. TISCH, K. SHOKAT, X. D. YANG, N. DUMONT, C. C. GOODNOW, AND H. O. MCDEVITT. Intravenous injection of soluble antigen induces thymic and peripheral T-cell apoptosis. Proc. Natl. Acad. Sci. USA 93: 3031–3036, 1996. LIN, H., S. F. BOLLING, P. S. LINSLEY, R.-Q. WEI, D. GORDON, C. B. THOMPSON, AND L. A. TURKA. Long-term acceptance of major histocompatibility complex mismatched cardiac allografts induced by CTLA4Ig plus donor-specific transfusion. J. Exp. Med. 178: 1801–1806, 1993. LIN, Y., M. VANDEPUTTE, AND M. WAER. Natural killer cell- and macrophage-mediated rejection of concordant xenografts in the absence of T and B cell responses. J. Immunol. 158: 5658–5667, 1997. LINSLEY, P. S., W. BRADY, L. GROSMAIRE, A. ARUFFO, N. K. DAMLE, AND J. A. LEDBETTER. Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J. Exp. Med. 173: 721–730, 1991. LINSLEY, P. S., W. BRADY, M. URNES, L. S. GROSMAIRE, N. K. DAMLE, AND J. A. LEDBETTER. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 174: 561–569, 1991. LINSLEY, P. S., J. L. GREENE, P. TAN, J. BRADSHAW, J. A. LEDBETTER, C. ANASETTI, AND N. K. DAMLE. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J. Exp. Med. 176: 1595–1604, 1992. LIU, Y. Is CTLA4 a negative regulator for T-cell activation? Immunol. Today 12: 569–572, 1997. LIU, Z., A. I. COLOVAI, S. TUGULEA, E. F. REED, P. E. FISHER, D. MANCINI, E. A. ROSE, R. CORTESINI, R. E. MICHLER, AND N. SUCIU-FOCA. Indirect recognition of donor HLA-DR peptides in organ allograft rejection. J. Clin. Invest. 98: 1150–1157, 1996. LIU, Z., Y. K. SUN, Y. P. XI, A. MAFFEI, E. REED, P. HARRIS, AND N. SUCIU-FOCA. Contribution of direct and indirect recognition pathways to T cell alloreactivity. J. Exp. Med. 177: 1643–1650, 1993. LOMBARDI, G., S. SIDHU, R. BATCHELOR, AND R. LECHLER. Anergic T cells as suppressor cells in vitro. Science 264: 1587–1589, 1994. LOWRY, R. P., T. TAKEUCHI, H. CREMISI, AND B. KONIECZNY. Th2-like effectors may function as antigen-specific suppressor cells in states of transplantation tolerance. Transplant. Proc. 25: 324– 326, 1993. LU, C. Y., T. A. KHAIR-EL-DIN, I. A. DAWIDSON, T. M. BUTLER, K. M. BRASKY, M. A. VAZQUEZ, AND S. C. SICHER. Xenotransplantation. FASEB J. 8: 1122–1130, 1994. LU, L., F. LI, F. G. CHAMBERS, S. QIAN, J. J. FUNG, AND A. W. THOMSON. Blockade of the CD40-CD40 ligand pathway potentiates the capacity of donor-derived dendritic cell progenitors to induce long-term cardiac allograft survival. Transplantation 64: 1808–1815, 1997. LU, L., S. QIAN, P. A. HERSHBERGER, W. A. RUDERT, D. H. LYNCH, AND A. W. THOMSON. Fas ligand (CD95L) and B7 expression on dendritic cells provide counter-regulatory signals for T cell survival and proliferation. J. Immunol. 158: 5676–5684, 1997. LUHDER, F., P. HOGLUND, J. P. ALLISON, C. BENOIST, AND D. MATHIS. Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) regulates the unfolding of autoimmune diabetes. J. Exp. Med. 187: 427–432, 1998. / 9j0d$$ja01 P26-8 135 232. LYNCH, D. H., M. L. WATSON, M. R. ALDERSON, P. R. BAUM, R. E. MILLER, T. TOUGH, M. GIBSON, T. DAVIS-SMITH, C. A. SMITH, K. HUNTER, D. BHAT, AND W. DIN. The mouse fas-ligand gene is mutated in gld mice and is part of a TNF family gene cluster. Immunity 1: 131–136, 1994. 233. MACDONALD, H. R., R. SCHNEIDER, R. K. LEES, R. C. HOWE, H. ACHA-ORBEA, H. FESTENSTEIN, R. M. ZINKERNAGEL, AND H. HENGARTNER. T-cell receptor V beta use predicts reactivity and tolerance to Mlsa-encoded antigens. Nature 332: 40–45, 1988. 234. MACH, F., U. SCHÖNBECK, G. K. SUKHOVA, T. BOURCIER, J. Y. BONNEFOY, J. S. POBER, AND P. LIBBY. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc. Natl. Acad. Sci. USA 94: 1931–1936, 1997. 235. MADRENAS, J., R. H. SCHWARTZ, AND R. N. GERMAIN. Interleukin 2 production, not the pattern of early T-cell antigen receptordependent tyrosine phosphorylation, controls anergy induction by both agonists and partial agonists. Proc. Natl. Acad. Sci. USA 93: 9736–9741, 1996. 236. MAEDA, H., M. TAKATA, S. TAKAHASHI, S. OGOSHI, AND S. FUJIMOTO. Adoptive transfer of a Th2-like cell line prolongs MHC class II antigen disparate skin allograft survival in the mouse. Int. Immunol. 6: 855–862, 1994. 236a.MAJNO, G. The Healing Hand. Man and Wound in the Ancient World. Cambridge, MA: Harvard Univ. Press, 1975, p. 261–312. 237. MANDEL, T. E. Fetal islet transplantation. Transplant. Proc. 24: 1996–1997, 1992. 238. MANDEL, T. E., AND M. KOULMANDA. Effect of anti-CD4 and antiCD8 monoclonal antibody treatment on fetal pig pancreas xenograft survival in nonobese diabetic/Lt female mice. Transplant. Proc. 24: 216–217, 1992. 239. MANDEL, T. E., M. KOULMANDA, E. COZZI, P. WATERWORTH, M. TOLAN, G. LANGFORD, AND D. J. G. WHITE. Transplantation of normal and DAF-transgenic fetal pig pancreas into cynomolgus monkeys. Transplant. Proc. 29: 940–940, 1997. 240. MARKEES, T. G., M. C. APPEL, R. J. NOELLE, J. P. MORDES, D. L. GREINER, AND A. A. ROSSINI. Tolerance to islet xenografts induced by dual manipulation of antigen presentation and co-stimulation. Transplant. Proc. 28: 814–815, 1996. 241. MARKEES, T. G., E. J. GORDON, N. E. PHILLIPS, L. D. SHULTZ, R. J. NOELLE, J. P. MORDES, D. L. GREINER, AND A. A. ROSSINI. Prolonged skin allo- and xenograft survival in C57BL/6 CD4-knockout mice treated with anti-CD154 and donor spleen cells. Transplant. Proc. In press. 242. MARKEES, T. G., N. E. PHILLIPS, E. J. GORDON, R. J. NOELLE, J. P. MORDES, D. L. GREINER, AND A. A. ROSSINI. Improved skin allograft tolerance induced by treatment with donor splenocytes and an extended course of anti-CD154 monoclonal antibody. Transplant. Proc. 30: 2444–2446, 1998. 242a.MARKEES, T. G., N. E. PHILLIPS, E. J. GORDON, R. J. NOELLE, J. P. MORDES, D. L. GREINER, AND A. A. ROSSINI. Prolonged skin allograft survival in mice treated with Flt3-ligand-induced dendritic cells and anti-CD154 monoclonal antibody. Transplant. Proc. In press. 243. MARKEES, T. G., N. E. PHILLIPS, E. J. GORDON, R. J. NOELLE, L. D. SHULTZ, J. P. MORDES, D. L. GREINER, AND A. A. ROSSINI. Long-term survival of skin allografts induced by donor splenocytes and anti-CD154 antibody in thymectomized mice requires CD4/ T cells, interferon-gamma, and CTLA4. J. Clin. Invest. 101: 2446– 2455, 1998. 244. MARKEES, T. G., N. E. PHILLIPS, R. J. NOELLE, L. D. SHULTZ, J. P. MORDES, D. L. GREINER, AND A. A. ROSSINI. Prolonged survival of mouse skin allografts in recipients treated with donor splenocytes and antibody to CD40 ligand. Transplantation 64: 329–335, 1997. 245. MARKMANN, J. F., J. TOMASZEWSKI, A. M. POSSELT, M. M. LEVY, M. WOEHRLE, C. F. BARKER, AND A. NAJI. The effect of islet cell culture in vitro at 247C on graft survival and MHC antigen expression. Transplantation 49: 272–277, 1990. 246. MARSHALL, S. E., S. P. COBBOLD, J. D. DAVIES, G. M. MARTIN, J. M. PHILLIPS, AND H. WALDMANN. Tolerance and suppression in a primed immune system. Transplantation 62: 1614–1621, 1996. 12-09-98 00:40:31 pra APS-Phys Rev 136 ROSSINI, GREINER, AND MORDES 247. MARUO, S., M. OH-HORA, H. J. AHN, S. ONO, M. WYSOCKA, Y. KANEKO, H. YAGITA, K. OKUMURA, H. KIKUTANI, T. KISHIMOTO, M. KOBAYASHI, AND T. HAMAOKA. B cells regulate CD40 ligand-induced IL-12 production in antigen-presenting cells (APC) during T cell/APC interactions. J. Immunol. 158: 120–126, 1997. 248. MATSUMURA, Y., A. MARCHEVSKY, X. J. ZUO, R. M. KASS, J. M. MATLOFF, AND S. C. JORDAN. Assessment of pathological changes associated with chronic allograft rejection and tolerance in two experimental models of rat lung transplantation. Transplantation 59: 1509–1517, 1995. 249. MATZINGER, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 12: 991–1045, 1994. 250. MAYO, G. L., A. M. POSSELT, C. F. BARKER, S. ROSTAMI, S. P. MAYO, L. CAMPOS, AND A. NAJI. Prolongation of survival of donorstrain islet xenografts (ratrmouse) by intrathymic inoculation of xenogeneic islet and bone marrow cells. Transplantation 58: 107– 109, 1994. 251. MCKENNEY, D. W., H. ONODERA, L. GORMAN, T. MIMURA, AND D. M. ROTHSTEIN. Distinct isoforms of the CD45 protein-tyrosine phosphatase differentially regulate interleukin 2 secretion and activation signal pathways involving Vav in T cells. J. Biol. Chem. 270: 24949–24954, 1995. 252. MERRILL, J. P. Transplantation immunology 1957–1975. Ann. Immunol. 129: 347–352, 1978. 253. MERRILL, J. P., J. E. MURRAY, J. H. HARRISON, AND W. R. GUILD. Landmark article Jan 28, 1956: successful homotransplantation of the human kidney between identical twins. JAMA 251: 2566–2571, 1984. 254. MILLER, J. F., AND W. R. HEATH. Self-ignorance in the peripheral T-cell pool. Immunol. Rev. 133: 131–150, 1993. 255. MILLER, R. G., S. MURAOKA, M. H. CLAESSON, J. REIMANN, AND P. BENVENISTE. The veto phenomenon in T-cell regulation. Ann. NY Acad. Sci. 532: 170–176, 1988. 256. MIWA, S., M. ISOBE, J. SUZUKI, M. MAKUUCHI, M. MIYASAKA, S. YAMAZAKI, AND S. KAWASAKI. Effect of anti-intercellular adhesion molecule-1 and anti-leukocyte function associated antigen-1 monoclonal antibodies on rat-to-mouse cardiac xenograft rejection. Surgery 121: 681–689, 1997. 257. MOGIL, R. J., L. RADVANYI, R. GONZALEZ-QUINTIAL, R. MILLER, G. MILLS, A. N. THEOFILOPOULOS, AND D. R. GREEN. Fas (CD95) participates in peripheral T cell deletion and associated apoptosis in vivo. Int. Immunol. 7: 1451–1458, 1995. 258. MOHLER, K. M., AND J. W. STREILEIN. Lymphokine production by MLR-reactive reaction lymphocytes obtained from normal mice and mice rendered tolerant of class II MHC antigens. Transplantation 47: 625–633, 1989. 259. MORDES, J. P., R. BORTELL, J. DOUKAS, M. R. RIGBY, B. J. WHALEN, D. ZIPRIS, D. L. GREINER, AND A. A. ROSSINI. The BB/ Wor rat and the balance hypothesis of autoimmunity. Diabetes Metab. Rev. 2: 103–109, 1996. 260. MORDES, J. P., D. L. GREINER, AND A. A. ROSSINI. Animal models of autoimmune diabetes mellitus. In: Diabetes Mellitus. A Fundamental and Clinical Text, edited by D. LeRoith, S. I. Taylor, and J. M. Olefsky. Philadelphia, PA: Lippincott-Raven, 1996, p. 349–360. 261. MORRIS, C. F., C. J. SIMEONOVIC, M. C. FUNG, J. D. WILSON, AND A. J. HAPEL. Intragraft expression of cytokine transcripts during pig proislet xenograft rejection and tolerance in mice. J. Immunol. 154: 2470–2482, 1995. 262. MORRIS, R. E. Investigational immunosuppressants: non-biologics. In: Primer on Transplantation, edited by D. J. Norman and W. N. Suki. Thorofare, NJ: Am. Soc. Transplant Physicians, 1998, p. 103– 112. 263. MOSKOPHIDIS, D., F. LECHNER, H. PIRCHER, AND R. M. ZINKERNAGEL. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 362: 758–761, 1993. 264. MOSMANN, T. R., AND S. SAD. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol. Today 17: 138–146, 1996. 265. MUELLER, D. L., M. K. JENKINS, AND R. H. SCHWARTZ. Clonal expansion versus functional clonal inactivation: a costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu. Rev. Immunol. 7: 445–480, 1989. 266. MURASE, N., T. E. STARZL, M. TANABE, S. FUJISAKI, H. MIYA- / 9j0d$$ja01 P26-8 267. 268. 269. 270. 271. 272. 273. 274. 275. 276. 277. 278. 279. 280. 281. 282. 283. 284. 285. 286. 287. 12-09-98 00:40:31 Volume 79 ZAWA, Q. YE, DELANEY, CP, J. J. FUNG, AND A. J. DEMETRIS. Variable chimerism, graft-versus-host disease, and tolerance after different kinds of cell and whole organ transplantation from Lewis to Brown Norway rats. Transplantation 60: 158–171, 1995. NAGATA, S., AND P. GOLSTEIN. The Fas death factor. Science 267: 1449–1456, 1995. NAGATA, S., AND T. SUDA. Fas and Fas ligand: lpr and gld mutations. Immunol. Today 16: 39–43, 1995. NASH, J. R., M. PETERS, AND P. R. F. BELL. Comparative survival of pancreatic islets, heart, kidney, and skin allografts in rats, with and without enhancement. Transplantation 24: 70–73, 1977. NELSON, C. A., S. J. PETZOLD, AND E. R. UNANUE. Peptides determine the lifespan of MHC class II molecules in the antigen-presenting cell. Nature 371: 250–252, 1994. NEVILLE, D. M., JR., J. SCHARFF, H. Z. HU, K. RIGAUT, J. SHILOACH, W. SLINGERLAND, AND M. JONKER. A new reagent for the induction of T-cell depletion, anti-CD3-CRM9. J. Immunother. 19: 85–92, 1996. NEWSTEAD, C. G. Assessment of risk of cancer after renal transplantation. Lancet 351: 610–611, 1998. NICKERSON, P., J. STEIGER, X. X. ZHENG, A. W. STEELE, W. STEURER, P. ROY-CHAUDHURY, AND T. B. STROM. Manipulation of cytokine networks in transplantation: false hope or realistic opportunity for tolerance? Transplantation 63: 489–494, 1997. NICKERSON, P., X. X. ZHENG, J. STEIGER, A. W. STEELE, W. STEURER, P. ROY-CHAUDHURY, W. MULLER, AND T. B. STROM. Prolonged islet allograft acceptance in the absence of interleukin 4 expression. Transplant Immunol. 4: 81–85, 1996. NIEDERKORN, J. Y. Immune privilege and immune regulation in the eye. Adv. Immunol. 48: 191–226, 1990. NIKOLIC, B., AND M. SYKES. Mixed hematopoietic chimerism and transplantation tolerance. Immunol. Res. 16: 217–228, 1997. NOELLE, R. J., J. A. LEDBETTER, AND A. ARUFFO. CD40 and its ligand, an essential ligand-receptor pair for thymus-dependent Bcell activation. Immunol. Today 13: 431–433, 1992. NOELLE, R. J., M. ROY, D. M. SHEPHERD, I. STAMENKOVIC, J. A. LEDBETTER, AND A. ARUFFO. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc. Natl. Acad. Sci. USA 89: 6550–6554, 1992. NORMAN, D. J. Expected clinical outcomes/risk factors. In: Primer on Transplantation, edited by D. J. Norman and W. N. Suki. Thorofare, NJ: Am. Soc. Transplant Physicians, 1998, p. 245–250. NOWAK, R. Xenotransplants set to resume. Science 266: 1148– 1151, 1994. ODORICO, J. S., C. F. BARKER, J. F. MARKMANN, A. M. POSSELT, AND A. NAJI. Prolonged survival of rat cardiac allografts after intrathymic inoculation of donor thymocytes. Transplant. Proc. 25: 295–296, 1993. ODORICO, J. S., C. F. BARKER, A. M. POSSELT, AND A. NAJI. Induction of donor-specific tolerance to rat cardiac allografts by intrathymic inoculation of bone marrow. Surgery 112: 370–377, 1992. ODORICO, J. S., A. M. POSSELT, A. NAJI, J. F. MARKMANN, AND C. F. BARKER. Promotion of rat cardiac allograft survival by intrathymic inoculation of donor splenocytes. Transplantation 55: 1104–1107, 1993. OHASHI, P. S., S. OEHEN, P. AICHELE, H. PIRCHER, B. ODERMATT, P. HERRERA, Y. HIGUCHI, K. BUERKI, H. HENGARTNER, AND R. M. ZINKERNAGEL. Induction of diabetes is influenced by the infectious virus and local expression of MHC class I and tumor necrosis factor-a. J. Immunol. 150: 5185–5194, 1993. OHASHI, P. S., S. OEHEN, K. BUERKI, H. PIRCHER, C. T. OHASHI, B. ODERMATT, B. MALISSEN, R. M. ZINKERNAGEL, AND H. HENGARTNER. Ablation of ‘‘tolerance’’ and induction of diabetes by virus infection in viral antigen transgenic mice. Cell 65: 305– 317, 1991. OHZATO, H., AND A. P. MONACO. Induction of specific unresponsiveness (tolerance) to skin allografts by intrathymic donor-specific splenocyte injection in antilymphocyte serum-treated mice. Transplantation 54: 1090–1095, 1992. OLUWOLE, S. F., N. C. CHOWDHURY, M. X. JIN, AND M. A. HARDY. Induction of transplantation tolerance to rat cardiac allo- pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION grafts by intrathymic inoculation of allogeneic soluble peptides. Transplantation 56: 1523–1527, 1993. 288. OLUWOLE, S. F., M. X. JIN, N. C. CHOWDHURY, AND O. A. OHAJEKWE. Effectiveness of intrathymic inoculation of soluble antigens in the induction of specific unresponsiveness to rat islet allografts without transient recipient immunosuppression. Transplantation 58: 1077–1081, 1994. 289. OLUWOLE, S. F., A. K. NG, K. REEMTSMA, AND M. A. HARDY. The mechanism of the induction of immunologic unresponsiveness to rat cardiac allografts by recipient pretreatment with donor lymphocyte subsets. Transplantation 48: 281–288, 1989. 290. OLUWOLE, S. F., K. REEMTSMA, AND M. A. HARDY. Characteristics and function of suppressor T lymphocytes in immunologically unresponsive rats following pretreatment with UV-B-irradiated donor leukocytes and peritransplant cyclosporine. Transplantation 47: 1001–1007, 1989. 291. ONODERA, H., D. G. MOTTO, G. A. KORETZKY, AND D. M. ROTHSTEIN. Differential regulation of activation-induced tyrosine phosphorylation and recruitment of SLP-76 to Vav by distinct isoforms of the CD45 protein-tyrosine phosphatase. J. Biol. Chem. 271: 22225–22230, 1996. 292. ONODERA, K., M. LEHMANN, E. AKALIN, H. D. VOLK, M. H. SAYEGH, AND J. W. KUPIEC-WEGLINSKI. Induction of ‘‘infectious’’ tolerance to MHC-incompatible cardiac allografts in CD4 monoclonal antibody-treated sensitized rat recipients. J. Immunol. 157: 1944– 1950, 1996. 293. OPELZ, G., AND P. I. TERASAKI. Poor kidney-transplant survival in recipients with frozen-blood transfusions or no transfusions. Lancet 2: 696–698, 1974. 294. ORLOFF, M. S., E. M. DEMARA, M. L. COPPAGE, N. LEONG, M. A. FALLON, J. SICKEL, X. J. ZUO, J. PREHN, AND S. C. JORDAN. Prevention of chronic rejection and graft arteriosclerosis by tolerance induction. Transplantation 59: 282–288, 1995. 294a.OWEN, R. D. Immunogenetic consequences of vascular anastomoses between bovine twins. Science 102: 400, 1945. 295. PARKER, D. C. T cell-dependent B cell activation. Annu. Rev. Immunol. 11: 331–360, 1993. 296. PARKER, D. C., AND E. E. EYNON. Antigen presentation in acquired immunological tolerance. FASEB J. 5: 2777–2784, 1991. 297. PARKER, D. C., D. L. GREINER, N. E. PHILLIPS, M. C. APPEL, A. W. STEELE, F. H. DURIE, R. J. NOELLE, J. P. MORDES, AND A. A. ROSSINI. Survival of mouse pancreatic islet allografts in recipients treated with allogeneic small lymphocytes and antibody to CD40 ligand. Proc. Natl. Acad. Sci. USA 92: 9560–9564, 1995. 298. PAUL, L. C. Immunobiology of chronic renal transplant rejection. Blood Purif. 13: 206–218, 1995. 299. PEARCE, N. W., A. SPINELLI, K. E. GURLEY, S. E. DORSCH, AND B. M. HALL. Mechanisms maintaining antibody-induced enhancement of allografts. II. Mediation of specific suppression by short lived CD4/ T cells. J. Immunol. 143: 499–506, 1989. 300. PEARSON, T. C., D. Z. ALEXANDER, M. CORBASCIO, R. HENDRIX, S. C. RITCHIE, P. S. LINSLEY, D. FAHERTY, AND C. P. LARSEN. Analysis of the B7 costimulatory pathway in allograft rejection. Transplantation 63: 1463–1469, 1997. 301. PEARSON, T. C., D. Z. ALEXANDER, R. HENDRIX, E. T. ELWOOD, P. S. LINSLEY, K. J. WINN, AND C. P. LARSEN. CTLA4-Ig plus bone marrow induces long-term allograft survival and donor specific unresponsiveness in the murine model. Evidence for hematopoietic chimerism. Transplantation 61: 997–1004, 1996. 302. PEARSON, T. C., D. Z. ALEXANDER, K. J. WINN, P. S. LINSLEY, R. P. LOWRY, AND C. P. LARSEN. Transplantation tolerance induced by CTLA4-Ig. Transplantation 57: 1701–1706, 1994. 303. PEGUET-NAVARRO, J., C. MOULON, C. CAUX, C. DALBIEZ-GAUTHIER, J. BANCHEREAU, AND D. SCHMITT. Interleukin-10 inhibits the primary allogeneic T cell response to human epidermal Langerhans cells. Eur. J. Immunol. 24: 884–891, 1994. 304. PENG, S. L., J. M. MCNIFF, M. P. MADAIO, J. A. MA, M. J. OWEN, R. A. FLAVELL, A. C. HAYDAY, AND J. CRAFT. ab T cell regulation and CD40 ligand dependence in murine systemic autoimmunity. J. Immunol. 158: 2464–2470, 1997. 305. PEREZ, V. L., L. VAN PARIJS, A. BIUCKIANS, X. X. ZHENG, T. B. STROM, AND A. K. ABBAS. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity 6: 411–417, 1997. / 9j0d$$ja01 P26-8 137 306. PHAM, S. M., R. J. KEENAN, A. S. RAO, P. A. FONTES, R. L. KORMOS, K. ABU-ELMAGD, A. ZEEVI, A. KAWAI, B. G. HATTLER, R. L. HARDESTY, A. J. DEMETRIS, AND M. M. TRUCCO. Perioperative donor bone marrow infusion augments chimerism in heart and lung transplant recipients. Ann. Thorac. Surg. 60: 1015–1020, 1995. 307. PICCOTTI, J. R., S. Y. CHAN, R. E. GOODMAN, J. MAGRAM, E. J. EICHWALD, AND D. K. BISHOP. IL-12 antagonism induces T helper 2 responses, yet exacerbates cardiac allograft rejection. Evidence against a dominant protective role for T helper 2 cytokines in alloimmunity. J. Immunol. 157: 1951–1957, 1996. 308. PICCOTTI, J. R., S. Y. CHAN, A. M. VANBUSKIRK, E. J. EICHWALD, AND D. K. BISHOP. Are Th2 helper T lymphocytes beneficial, deleterious, or irrelevant in promoting allograft survival? Transplantation 63: 619–624, 1997. 309. PLAIN, K. M., L. FAVA, A. SPINELLI, X. Y. HE, J. CHEN, R. BOYD, C. L. DAVIDSON, AND B. M. HALL. Induction of tolerance with nondepleting anti-CD4 monoclonal antibodies is associated with downregulation of Th2 cytokines. Transplantation 64: 1559–1567, 1997. 310. POSSELT, A. M., C. F. BARKER, A. L. FRIEDMAN, B. KOEBERLEIN, J. E. TOMASZEWSKI, AND A. NAJI. Intrathymic inoculation of islets at birth prevents autoimmune diabetes and pancreatic insulitis in the BB rat. Transplant. Proc. 25: 301–302, 1993. 311. POSSELT, A. M., C. F. BARKER, J. E. TOMASZEWSKI, J. F. MARKMANN, M. A. CHOTI, AND A. NAJI. Induction of donor-specific unresponsiveness by intrathymic islet transplantation. Science 249: 1293–1295, 1990. 312. POWELL, T. J., JR., AND J. W. STREILEIN. Neonatal tolerance induction by class II alloantigens activates IL-4-secreting, tolerogenresponsive T cells. J. Immunol. 144: 854–859, 1990. 313. PRIESTLEY, C. A., S. C. SPENCER, G. J. SAWYER, AND J. W. FABRE. Suppression of kidney allograft rejection across full MHC barriers by recipient-specific antibodies to class II MHC antigens. Transplantation 53: 1024–1032, 1992. 314. QIN, S., S. P. COBBOLD, H. POPE, J. ELLIOTT, D. KIOUSSIS, J. DAVIES, AND H. WALDMANN. ‘‘Infectious’’ transplantation tolerance. Science 259: 974–977, 1993. 315. QUILL, H. Anergy as a mechanism of peripheral T cell tolerance. J. Immunol. 156: 1325–1327, 1996. 316. RAJU, G. P., S. E. BELLAND, AND H. J. EISEN. Prolongation of cardiac allograft survival with transforming growth factor-beta 1 in rats. Transplantation 58: 392–396, 1994. 317. RAMSDELL, F., AND B. J. FOWLKES. Clonal deletion versus clonal anergy: the role of the thymus in inducing self tolerance. Science 248: 1342–1348, 1990. 318. RAMSDELL, F., T. LANTZ, AND B. J. FOWLKES. A nondeletional mechanism of thymic self tolerance. Science 246: 1038–1041, 1989. 319. RANHEIM, E. A., AND T. J. KIPPS. Activated T cells induce expression of B7/BB1 on normal or leukemic B cells through a CD40dependent signal. J. Exp. Med. 177: 925–935, 1993. 320. RÄISÄNEN-SOKOLOWSKI, A., P. L. MOTTRAM, T. GLYSING-JENSEN, A. SATOSKAR, AND M. E. RUSSELL. Heart transplants in interferon-gamma, interleukin 4, and interleukin 10 knockout mice: recipient environment alters graft rejection. J. Clin. Invest. 100: 2449–2456, 1997. 321. REICH, E.-P., D. SCARINGE, J. YAGI, R. S. SHERWIN, AND C. A. JANEWAY, JR. Prevention of diabetes in NOD mice by injection of autoreactive T-lymphocytes. Diabetes 38: 1647–1651, 1989. 322. REINSMOEN, N. L., A. JACKSON, C. MCSHERRY, D. NINOVA, R. H. WIESNER, M. KONDO, R. A. KROM, M. I. HERTZ, R. M. BOLMAN III, AND A. J. MATAS. Organ-specific patterns of donor antigenspecific hyporeactivity and peripheral blood allogeneic microchimerism in lung, kidney, and liver transplant recipients. Transplantation 60: 1546–1554, 1995. 323. REINSMOEN, N. L., AND A. J. MATAS. Evidence that improved late renal transplant outcome correlates with the development of in vitro donor antigen-specific hyporeactivity. Transplantation 55: 1017–1023, 1993. 324. REMUZZI, G., M. NORIS, A. BENIGNI, O. IMBERTI, M. H. SAYEGH, AND N. PERICO. Thromboxane A2 receptor blocking abrogates donor-specific unresponsiveness to renal allografts induced by thymic recognition of major histocompatibility allopeptides. J. Exp. Med. 180: 1967–1972, 1994. 325. REMUZZI, G., M. ROSSINI, O. IMBERTI, AND N. PERICO. Kidney 12-09-98 00:40:31 pra APS-Phys Rev 138 326. 327. 328. 329. 330. 331. 332. 333. 334. 335. 336. 337. 338. 339. 340. 341. 342. 343. 344. 345. 346. 347. 348. ROSSINI, GREINER, AND MORDES graft survival in rats without immunosuppressants after intrathymic glomerular transplantation. Lancet 337: 750–752, 1991. REP, M. H., B. W. VAN OOSTEN, M. T. ROOS, H. J. ADER, C. H. POLMAN, AND R. A. VAN LIER. Treatment with depleting CD4 monoclonal antibody results in a preferential loss of circulating naive T cells but does not affect IFN-gamma secreting TH1 cells in humans. J. Clin. Invest. 99: 2225–2231, 1997. RIDGWAY, W. M., H. L. WEINER, AND C. G. FATHMAN. Regulation of autoimmune response. Curr. Opin. Immunol. 6: 946–955, 1994. ROBSON, S. C., D. CANDINAS, W. W. HANCOCK, C. WRIGHTON, H. WINKLER, AND F. H. BACH. Role of endothelial cells in transplantation. Int. Arch. Allergy Immunol. 106: 305–322, 1995. ROCHA, B., A. GRANDIEN, AND A. A. FREITAS. Anergy and exhaustion are independent mechanisms of peripheral T cell tolerance. J. Exp. Med. 181: 993–1003, 1995. ROCHA, B., AND H. VON BOEHMER. Peripheral selection of the T cell repertoire. Science 251: 1225–1228, 1991. ROCK, K. L., AND K. CLARK. Analysis of the role of MHC class II presentation in the stimulation of cytotoxic T lymphocytes by antigens targeted into the exogenous antigen-MHC class I presentation pathway. J. Immunol. 156: 3721–3726, 1996. ROGGE, L., L. BARBERIS-MAINO, M. BIFFI, N. PASSINI, D. H. PRESKY, U. GUBLER, AND F. SINIGAGLIA. Selective expression of an interleukin-12 receptor component by human T helper 1 cells. J. Exp. Med. 185: 825–831, 1997. ROSENBERG, A. S., AND A. SINGER. Cellular basis of skin allograft rejection: an in vivo model of immune-mediated tissue destruction. Annu. Rev. Immunol. 10: 333–358, 1992. ROSER, B. J. Cellular mechanisms in neonatal and adult tolerance. Immunol. Rev. 107: 179–202, 1989. ROSSINI, A. A., J. P. MORDES, E. S. HANDLER, AND D. L. GREINER. Human autoimmune diabetes mellitus: lessons from BB rats and NOD mice—Caveat emptor. Clin. Immunol. Immunopathol. 74: 2–9, 1995. ROSSINI, A. A., J. P. MORDES, T. G. MARKEES, N. E. PHILLIPS, E. J. GORDON, AND D. L. GREINER. Induction of islet transplantation tolerance using donor specific transfusion and anti-CD154 monoclonal antibody. Transplant. Proc. In press. ROSSINI, A. A., D. C. PARKER, N. E. PHILLIPS, F. H. DURIE, R. J. NOELLE, J. P. MORDES, AND D. L. GREINER. Induction of immunological tolerance to islet grafts. Cell Transplant. 5: 49–52, 1996. ROUSH, W. Xenotransplantation. New ways to avoid organ rejection buoy hopes. Science 234–235, 1995. ROUVIER, E., M. F. LUCIANI, AND P. GOLSTEIN. Fas involvement in Ca2/-independent T cell-mediated cytotoxicity. J. Exp. Med. 177: 195–200, 1993. ROY-CHAUDHURY, P., P. W. NICKERSON, R. C. MANFRO, X. X. ZHENG, J. STEIGER, Y. S. LI, AND T. B. STROM. CTLA4Ig attenuates accelerated rejection (presensitization) in the mouse islet allograft model. Transplantation 64: 172–175, 1997. SABLINSKI, T., W. W. HANCOCK, N. L. TILNEY, AND J. W. KUPIECWEGLINSKI. CD4 monoclonal antibodies in organ transplantation—a review of progress. Transplantation 52: 579–589, 1991. SACHS, D. H. Specific transplantation tolerance. N. Engl. J. Med. 325: 1240–1242, 1991. SACHS, D. H., AND F. H. BACH. Immunology of xenograft rejection. Hum. Immunol. 28: 245–251, 1990. SAMOILOVA, E. B., J. L. HORTON, H. D. ZHANG, AND Y. H. CHEN. CD40L blockade prevents autoimmune encephalomyelitis and hampers TH1 but not TH2 pathway of T cell differentiation. J. Mol. Med. 75: 603–608, 1997. SAYEGH, M. H., E. AKALIN, W. W. HANCOCK, M. E. RUSSELL, C. B. CARPENTER, P. S. LINSLEY, AND L. A. TURKA. CD28-B7 blockade after alloantigenic challenge in vivo inhibits Th1 cytokines but spares Th2. J. Exp. Med. 181: 1869–1874, 1995. SAYEGH, M. H., AND C. B. CARPENTER. Role of indirect allorecognition in allograft rejection. Int. Rev. Immunol. 13: 221–229, 1996. SAYEGH, M. H., S. J. KHOURY, W. W. HANCOCK, H. L. WEINER, AND C. B. CARPENTER. Induction of immunity and oral tolerance with polymorphic class II major histocompatibility complex allopeptides in the rat. Proc. Natl. Acad. Sci. USA 89: 7762–7766, 1992. SAYEGH, M. H., N. PERICO, O. IMBERTI, W. W. HANCOCK, C. B. CARPENTER, AND G. REMUZZI. Thymic recognition of class II / 9j0d$$ja01 P26-8 349. 350. 351. 352. 353. 354. 355. 356. 357. 358. 359. 360. 361. 362. 363. 364. 365. 366. 367. 368. 369. 370. 12-09-98 00:40:31 Volume 79 major histocompatibility complex allopeptides induces donor-specific unresponsiveness to renal allografts. Transplantation 56: 461– 465, 1993. SAYEGH, M. H., B. WATSCHINGER, AND C. B. CARPENTER. Mechanisms of T cell recognition of alloantigen. The role of peptides. Transplantation 57: 1295–1302, 1994. SAYEGH, M. H., X. G. ZHENG, C. MAGEE, W. W. HANCOCK, AND L. A. TURKA. Donor antigen is necessary for the prevention of chronic rejection in CTLA4Ig-treated murine cardiac allograft recipients. Transplantation 64: 1646–1650, 1997. SCHAUB, M., T. H. W. STADLBAUER, AND M. H. SAYEGH. CTLA4Ig: effects on cellular and humoral immunity and macrophage activation. Exp. Nephrol. 5: 370–374, 1997. SCHONRICH, G., U. KALINKE, F. MOMBURG, M. MALISSEN, A. M. SCHMITT-VERHULST, B. MALISSEN, G. J. HAMMERLING, AND B. ARNOLD. Down-regulation of T cell receptors on self-reactive T cells as a novel mechanism for extrathymic tolerance induction. Cell 65: 293–304, 1991. SCHONRICH, G., F. MOMBURG, G. J. HAMMERLING, AND B. ARNOLD. Anergy induced by thymic medullary epithelium. Eur. J. Immunol. 22: 1687–1691, 1992. SCHREINER, G. F., W. FLYE, E. BRUNT, K. KORBER, AND J. B. LEFKOWITH. Essential fatty acid depletion of renal allografts and prevention of rejection. Science 240: 1032–1033, 1988. SCHWARTZ, R. H. Acquisition of immunological self-tolerance. Cell 57: 1073–1081, 1989. SCHWARTZ, R. H. A cell culture model for T lymphocyte clonal anergy. Science 248: 1349–1356, 1990. SCHWARTZ, R. H. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell 71: 1065–1068, 1992. SCHWARTZ, R. H. Models of T cell anergy: is there a common molecular mechanism? J. Exp. Med. 184: 1–8, 1996. SCOTT, B., J. KAYE, AND D. LO. T cells and suppression in vitro. Science 266: 464–464, 1994. SCOTT, D. M., I. E. EHRMANN, P. S. ELLIS, C. E. BISHOP, A. I. AGULNIK, E. SIMPSON, AND M. J. MITCHELL. Identification of a mouse male-specific transplantation antigen, H-Y. Nature 376: 695– 698, 1995. SCULLY, R., S. P. COBBOLD, A. L. MELLOR, M. WISSING, B. ARNOLD, AND H. WALDMANN. A role for Th2 cytokines in the suppression of CD8/ T cell-mediated graft rejection. Eur. J. Immunol. 27: 1663–1670, 1997. SCULLY, R., S. QIN, S. COBBOLD, AND H. WALDMANN. Mechanisms in CD4 antibody-mediated transplantation tolerance: kinetics of induction, antigen dependency and role of regulatory T cells. Eur. J. Immunol. 24: 2383–2392, 1994. SEINO, K., N. KAYAGAKI, N. TSUKADA, K. FUKAO, H. YAGITA, AND K. OKUMURA. Transplantation of CD95 ligand-expressing grafts: influence of transplantation site and difficulty in protecting allo- and xenografts. Transplantation 64: 1050–1054, 1997. SELAWRY, H. P., AND D. F. CAMERON. Sertoli cell-enriched fractions in successful islet cell transplantation. Cell Transplant. 2: 123–129, 1993. SEYDEL, K., J. SHIZURU, D. GROSSMAN, A. WU, S. ALTERS, AND C. G. FATHMAN. Anti-CD8 abrogates effect of anti-CD4-mediated islet allograft survival in rat model. Diabetes 40: 1430–1434, 1991. SHARMA, V. K., B. LI, A. KHANNA, P. K. SEHAJPAL, AND M. SUTHANTHIRAN. Which way for drug-mediated immunosuppression? Curr. Opin. Immunol. 6: 784–790, 1994. SHIN, Y. T., D. H. ADAMS, L. R. WYNER, E. AKALIN, M. H. SAYEGH, AND M. J. KARNOVSKY. Intrathymic tolerance in the Lewisto-F344 chronic cardiac allograft rejection model. Transplantation 59: 1647–1653, 1995. SHIZURU, J. A., S. E. ALTERS, AND C. G. FATHMAN. Anti-CD4 monoclonal antibodies in therapy: creation of nonclassical tolerance in the adult. Immunol. Rev. 129: 105–130, 1992. SHIZURU, J. A., A. K. GREGORY, C. T. CHAO, AND C. G. FATHMAN. Islet allograft survival after a single course of treatment of recipient with antibody to L3T4. Science 237: 278–280, 1987. SIEGLING, A., M. LEHMANN, H. RIEDEL, C. PLATZER, J. BROCK, F. EMMRICH, AND H. D. VOLK. A nondepleting anti-rat CD4 monoclonal antibody that suppresses T helper 1-like but not T helper 2- pra APS-Phys Rev January 1999 371. 372. 373. 374. 375. 376. 377. 378. 379. 380. 381. 382. 383. 384. 385. 386. 387. 388. 389. 390. 391. INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION like intragraft lymphokine secretion induces long-term survival of renal allografts. Transplantation 57: 464–467, 1994. SILVERSTEIN, A. M. A History of Immunology. San Diego, CA: Academic, 1988, p. 1–422. SIMEONOVIC, C. J., S. J. PROWSE, AND K. J. LAFFERTY. Reversal of diabetes in outbred mice by islet allotransplantation. Diabetes 35: 1345–1349, 1986. SIMEONOVIC, C. J., AND J. D. WILSON. Islets and tolerance. In: Pancreatic Islet Cell Transplantation: 1892–1992, One Century of Transplantation for Diabetes, edited by C. Ricordi. Austin, TX: Landes, 1992, p. 383–393. SINGER, G. G., AND A. K. ABBAS. The fas antigen is involved in peripheral but not thymic deletion of T lymphocytes in T cell receptor transgenic mice. Immunity 1: 365–371, 1994. SMITH, P. A., A. BRUNMARK, M. R. JACKSON, AND T. A. POTTER. Peptide-independent recognition by alloreactive cytotoxic T lymphocytes (CTL). J. Exp. Med. 185: 1023–1033, 1997. SMITH, R. M., Z. K. CHEN, R. FOULKES, S. M. METCALFE, AND D. C. WRAITH. Prolongation of murine vascularized heart allograft survival by recipient-specific anti-major histocompatibility complex class II antibody. Transplantation 64: 525–528, 1997. SMYTH, M. J. Fas ligand-mediated bystander lysis of syngeneic cells in response to an allogeneic stimulus. J. Immunol. 158: 5765– 5772, 1997. SONG, H. K., S. E. ALTERS, AND C. G. FATHMAN. Evidence that anti-CD8 abrogates anti-CD4-mediated clonal anergy but allows allograft survival in mice. Transplantation 55: 133–139, 1993. SOULILLOU, J. P., F. BLANDIN, E. GUNTHER, AND V. LEMOINE. Genetics of the blood transfusion effect on heart allografts in rats. Transplantation 38: 63–67, 1984. SPRENT, J., M. HURD, M. SCHAEFER, AND W. HEATH. Split tolerance in spleen chimeras. J. Immunol. 154: 1198–1206, 1995. SPRIGGS, M. K., R. J. ARMITAGE, L. STROCKBINE, K. N. CLIFFORD, B. M. MACDUFF, T. A. SATO, C. R. MALISZEWSKI, AND W. C. FANSLOW. Recombinant human CD40 ligand stimulates B cell proliferation and immunoglobulin E secretion. J. Exp. Med. 176: 1543–1550, 1992. STADLBAUER, T. H. W., M. SCHAUB, S. KOROM, K. ONODERA, M. H. SAYEGH, AND J. W. KUPIEC-WEGLINSKI. CD28-B7 T-cell costimulatory blockade potentiates the effects of intrathymic immunomodulation in sensitized graft recipients. Transplantation 64: 1816–1822, 1997. STAPLES, P. J., I. GERY, AND B. H. WAKSMAN. Role of the thymus in tolerance. 3. Tolerance to bovine gamma globulin after direct injection of antigen into the shielded thymus of irradiated rats. J. Exp. Med. 124: 127–139, 1966. STARZL, T. E., A. J. DEMETRIS, N. MURASE, A. W. THOMSON, M. TRUCCO, AND C. RICORDI. Donor cell chimerism permitted by immunosuppressive drugs: a new view of organ transplantation. Trends Pharmacol. Sci. 14: 217–223, 1993. STARZL, T. E., A. J. DEMETRIS, N. MURASE, M. TRUCCO, A. W. THOMSON, AND A. S. RAO. The lost chord: microchimerism and allograft survival. Immunol. Today 17: 577–584, 1996. STATTER, M. B., K. J. FAHRNER, E. M. BARKSDALE, JR., D. E. PARKS, R. A. FLAVELL, AND P. K. DONAHOE. Correlation of fetal kidney and testis congenic graft survival with reduced major histocompatibility complex burden. Transplantation 47: 651–660, 1989. STATTER, M. B., R. P. FOGLIA, D. E. PARKS, AND P. K. DONAHOE. Fetal and postnatal testis shows immunoprivilege as donor tissue. J. Urol. 139: 204–210, 1988. STEIGER, J., P. W. NICKERSON, W. STEURER, M. MOSCOVITCHLOPATIN, AND T. B. STROM. IL-2 knockout recipient mice reject islet cell allografts. J. Immunol. 155: 489–498, 1995. STEINMULLER, D. Passenger leukocytes and the immunogenicity of skin allografts: a critical reevaluation. Transplant. Proc. 13: 1094–1098, 1981. STEURER, W., P. W. NICKERSON, A. W. STEELE, J. STEIGER, X. X. ZHENG, AND T. B. STROM. Ex vivo coating of islet cell allografts with murine CTLA4/Fc promotes graft tolerance. J. Immunol. 155: 1165–1174, 1995. STREILEIN, J. W., B. R. KSANDER, AND A. W. TAYLOR. Immune deviation in relation to ocular immune privilege. J. Immunol. 158: 3557–3560, 1997. / 9j0d$$ja01 P26-8 139 392. STREILEIN, J. W., M. TAKEUCHI, AND A. W. TAYLOR. Immune privilege, T-cell tolerance, and tissue-restricted autoimmunity. Hum. Immunol. 52: 138–143, 1997. 393. STROM, T. B., AND R. B. ETTENGER. Investigational immunosuppressants: biologics. In: Primer on Transplantation, edited by D. J. Norman and W. N. Suki. Thorofare, NJ: Am. Soc. Transplant Physicians, 1998, p. 113–122. 394. STUART, P. M., T. S. GRIFFITH, N. USUI, J. PEPOSE, X. H. YU, AND T. A. FERGUSON. CD95 ligand (FasL)-induced apoptosis is necessary for corneal allograft survival. J. Clin. Invest. 99: 396– 402, 1997. 395. STÜBER, E., W. STROBER, AND M. NEURATH. Blocking the CD40L-CD40 interaction in vivo specifically prevents the priming of T helper 1 cells through the inhibition of interleukin 12 secretion. J. Exp. Med. 183: 693–698, 1996. 396. SUBBOTIN, V., H. SUN, A. AITOUCHE, L. A. VALDIVIA, J. J. FUNG, T. E. STARZL, AND A. S. RAO. Abrogation of chronic rejection in a murine model of aortic allotransplantation by prior induction of donor-specific tolerance. Transplantation 64: 690–695, 1997. 397. SUBERBIELLE, C., S. CAILLAT-ZUCMAN, C. LEGENDRE, C. BODEMER, L. H. NOEL, H. KREIS, AND J. F. BACH. Peripheral microchimerism in long-term cadaveric-kidney allograft recipients. Lancet 343: 1468–1469, 1994. 398. SUCIU-FOCA, N., E. REED, V. D. D’AGATI, E. HO, D. J. COHEN, A. I. BENVENISTY, R. MCCABE, J. M. BRENSILVER, D. W. KING, AND M. A. HARDY. Soluble HLA antigens, anti-HLA antibodies, and anti-idiotypic antibodies in the circulation of renal transplant recipients. Transplantation 51: 593–601, 1991. 399. SUN, H., V. SUBBOTIN, C. CHEN, A. AITOUCHE, L. A. VALDIVIA, M. H. SAYEGH, P. S. LINSLEY, J. J. FUNG, T. E. STARZL, AND A. S. RAO. Prevention of chronic rejection in mouse aortic allografts by combined treatment with CTLA4-Ig and anti-CD40 ligand monoclonal antibody. Transplantation 64: 1838–1843, 1997. 400. SUSS, G., AND K. SHORTMAN. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand-induced apoptosis. J. Exp. Med. 183: 1789–1796, 1996. 401. SUTHERLAND, D. E., R. SIBLEY, X. Z. XU, A. MICHAEL, A. M. SRIKANTA, F. TAUB, J. NAJARIAN, AND F. C. GOETZ. Twin-totwin pancreas transplantation: reversal and reenactment of the pathogenesis of type I diabetes. Trans. Assoc. Am. Physicians 97: 80–87, 1984. 402. SWAIN, S. L. T-cell subsets. Who does the polarizing? Curr. Biol. 5: 849–851, 1995. 403. SWAIN, S. L., M. CROFT, C. DUBEY, L. HAYNES, P. ROGERS, X. ZHANG, AND L. M. BRADLEY. From naive to memory T cells. Immunol. Rev. 150: 143–167, 1996. 404. SWAT, W., L. IGNATOWICZ, H. VON BOEHMER, AND P. KISIELOW. Clonal deletion of immature CD4/8/ thymocytes in suspension culture by extrathymic antigen-presenting cells. Nature 351: 150–153, 1991. 405. SYKES, M. Immunobiology of transplantation. FASEB J. 10: 721– 730, 1996. 406. SZABO, S. J., A. S. DIGHE, U. GUBLER, AND K. M. MURPHY. Regulation of the interleukin (IL)-12R beta 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J. Exp. Med. 185: 817–824, 1997. 407. TAGA, K., H. MOSTOWSKI, AND G. TOSATO. Human interleukin10 can directly inhibit T-cell growth. Blood 81: 2964–2971, 1993. 408. TAKAHASHI, T., M. TANAKA, C. I. BRANNAN, N. A. JENKINS, N. G. COPELAND, T. SUDA, AND S. NAGATA. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell 76: 969–976, 1994. 409. TAN, P., C. ANASETTI, J. A. HANSEN, J. MELROSE, M. BRUNVAND, J. BRADSHAW, J. A. LEDBETTER, AND P. S. LINSLEY. Induction of alloantigen-specific hyporesponsiveness in human T lymphocytes by blocking interaction of CD28 with its natural ligand B7/BB1. J. Exp. Med. 177: 165–173, 1993. 410. TERAOKA, S., T. KAWAI, AND Y. YAMAGUCHI. Mechanism of the preventive effect of aminobenzoic acid salt on pancreatic beta cell toxicity by cyclosporin. Transplant. Proc. 22: 863–865, 1990. 411. TERASAKA, R., P. E. LACY, V. HAUPTFELD, R. P. BUCY, AND J. M. DAVIE. The effect of cyclosporin-A, low-temperature culture, and 12-09-98 00:40:31 pra APS-Phys Rev 140 412. 413. 414. 415. 416. 417. 418. 419. 420. 421. 422. 423. 424. 425. 426. 427. 428. 429. 430. 431. ROSSINI, GREINER, AND MORDES anti-Ia antibodies on prevention of rejection of rat islet allografts. Diabetes 35: 83–88, 1986. THOMAS, J. M., K. M. VERBANAC, J. P. SMITH, J. KASTEN-JOLLY, U. GROSS, L. M. REBELLATO, C. E. HAISCH, F. M. CARVER, AND F. T. THOMAS. The facilitating effect of one-DR antigen sharing in renal allograft tolerance induced by donor bone marrow in rhesus monkeys. Transplantation 59: 245–255, 1995. THOMPSON, C. B., AND J. P. ALLISON. The emerging role of CTLA4 as an immune attenuator. Immunity 7: 445–450, 1997. THOMPSON, C. B., T. LINDSTEN, J. A. LEDBETTER, S. L. KUNKEL, H. A. YOUNG, S. G. EMERSON, J. M. LEIDEN, AND C. H. JUNE. CD28 activation pathway regulates the production of multiple T-cell-derived lymphokines/cytokines. Proc. Natl. Acad. Sci. USA 86: 1333–1337, 1989. TIVOL, E. A., F. BORRIELLO, A. N. SCHWEITZER, W. P. LYNCH, J. A. BLUESTONE, AND A. H. SHARPE. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction. Revealing a critical negative regulatory role of CTLA-4. Immunity 3: 541–547, 1995. TIVOL, E. A., S. D. BOYD, S. MCKEON, F. BORRIELLO, P. NICKERSON, T. B. STROM, AND A. H. SHARPE. CTLA4Ig prevents lymphoproliferation and fatal multiorgan tissue destruction in CTLA-4deficient mice. J. Immunol. 158: 5091–5094, 1997. TIVOL, E. A., A. N. SCHWEITZER, AND A. H. SHARPE. Costimulation and autoimmunity. Curr. Opin. Immunol. 8: 822–830, 1996. TRAN, H. M., P. W. NICKERSON, A. C. RESTIFO, M. A. IVISWOODWARD, A. PATEL, R. D. ALLEN, T. B. STROM, AND P. J. O’CONNELL. Distinct mechanisms for the induction and maintenance of allograft tolerance with CTLA4-Fc treatment. J. Immunol. 159: 2232–2239, 1997. TROWBRIDGE, I. S., AND M. L. THOMAS. CD45: an emerging role as a protein tyrosine phosphatase required for lymphocyte activation and development. Annu. Rev. Immunol. 12: 85–116, 1994. TRUCCO, M., AND G. STASSI. Transplantation biology: educating effector T cells. Nature 380: 284–285, 1996. TUCH, B. E., J. BERETOV, T. E. MANDEL, AND M. KOULMANDA. Effect of anti-CD4 monoclonal antibody on survival of xenografted human fetal pancreas. Transplant. Proc. 27: 2183–2184, 1995. TURKA, L. A., P. S. LINSLEY, H. LIN, W. BRADY, J. M. LEIDEN, R. Q. WEI, M. L. GIBSON, X. G. ZHENG, S. MYRDAL, AND D. GORDON. T-cell activation by the CD28 ligand B7 is required for cardiac allograft rejection in vivo. Proc. Natl. Acad. Sci. USA 89: 11102– 11105, 1992. VANBUSKIRK, A. M., M. E. WAKELY, AND C. G. OROSZ. Transfusion of polarized TH2-like cell populations into SCID mouse cardiac allograft recipients results in acute allograft rejection. Transplantation 62: 229–238, 1996. VANHOVE, B., AND F. H. BACH. Human xenoreactive natural antibodies—avidity and targets on porcine endothelial cells. Transplantation 56: 1251–1253, 1993. VAN KOOTEN, C., AND J. BANCHEREAU. Functional role of CD40 and its ligand. Int. Arch. Allergy Immunol. 113: 393–399, 1997. VAN TWUYVER, E., W. M. KAST, R. J. MOOIJAART, C. J. MELIEF, AND L. P. DE WAAL. Induction of transplantation tolerance by intravenous injection of allogeneic lymphocytes across an H-2 class II mismatch. Different mechanisms operate in tolerization across an H-2 class I vs. H-2 class II disparity. Eur. J. Immunol. 20: 441–444, 1990. VOJTISKOVA, M., AND A. LENGEROVA. Thymus-mediated tolerance to cellular alloantigens. Transplantation 6: 13–24, 1968. VOSSEN, A. C., G. J. TIBBE, R. BENNER, AND H. F. SAVELKOUL. T-lymphocyte and cytokine-directed strategies for inhibiting skin allograft rejection in mice. Transplant. Proc. 27: 380–382, 1995. WAANDERS, G. A., A. N. SHAKHOV, W. HELD, O. KARAPETIAN, H. ACHA-ORBEA, AND H. R. MACDONALD. Peripheral T cell activation and deletion induced by transfer of lymphocyte subsets expressing endogenous or exogenous mouse mammary tumor virus. J. Exp. Med. 177: 1359–1366, 1993. WALTENBERGER, J., A. WANDERS, B. FELLSTROM, K. MIYAZONO, C. H. HELDIN, AND K. FUNA. Induction of transforming growth factor-beta during cardiac allograft rejection. J. Immunol. 151: 1147–1157, 1993. WANG, Z. Q., A. DUDHANE, T. ORLIKOWSKY, K. CLARKE, X. LI, / 9j0d$$ja01 P26-8 432. 433. 434. 435. 436. 437. 438. 439. 440. 441. 442. 443. 444. 445. 446. 447. 448. 449. 450. 451. 452. 453. 12-09-98 00:40:31 Volume 79 Z. DARZYNKIEWICZ, AND M. K. HOFFMANN. CD4 engagement induces Fas antigen-dependent apoptosis of T cells in vivo. Eur. J. Immunol. 24: 1549–1552, 1994. WARRENS, A. N., G. LOMBARDI, AND R. I. LECHLER. Presentation and recognition of major and minor histocompatibility antigens. Transplant Immunol. 2: 103–107, 1994. WATANABE-FUKUNAGA, R., C. I. BRANNAN, N. G. COPELAND, N. A. JENKINS, AND S. NAGATA. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 356: 314–317, 1992. WATERHOUSE, P., J. M. PENNINGER, E. TIMMS, A. WAKEHAM, A. SHAHINIAN, K. P. LEE, C. B. THOMPSON, H. GRIESSER, AND T. W. MAK. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270: 985–988, 1995. WEBB, S., C. MORRIS, AND J. SPRENT. Extrathymic tolerance of mature T cells: clonal elimination as a consequence of immunity. Cell 63: 1249–1256, 1990. WEIGLE, W. O., AND C. G. ROMBALL. T-cell subsets and cytokines involved in peripheral tolerance. Immunol. Today 18: 533–538, 1997. WEINER, H. L. Oral tolerance for the treatment of autoimmune diseases. Annu. Rev. Med. 48: 341–351, 1997. WEINER, H. L. Oral tolerance: immune mechanisms and treatment of autoimmune diseases. Immunol. Today 18: 335–343, 1997. WEINER, H. L., A. MILLER, S. J. KHOURY, Z. J. ZHANG, A. ALSABBAGH, S. A. BROD, O. LIDER, P. HIGGINS, R. SOBEL, M. MATSUI, M. SAYEGH, AND C. CARPENTER. Treatment of autoimmune diseases by oral tolerance to autoantigens. Adv. Exp. Med. Biol. 371: 1217–1223, 1995. WEKERLE, T., M. H. SAYEGH, J. HILL, Y. ZHAO, A. CHANDRAKER, K. G. SWENSON, G. ZHAO, AND M. SYKES. Extrathymic T cell deletion and allogeneic stem cell engraftment induced with costimulatory blockade is followed by central T cell tolerance. J. Exp. Med. 187: 2037–2044, 1998. WHITE, D. J. G., AND N. YANNOUTSOS. Production of pigs transgenic for human DAF to overcome complement-mediated hyperacute xenograft rejection in man. Res. Immunol. 147: 88–94, 1996. WILLEMS, F., A. MARCHANT, J. P. DELVILLE, C. GERARD, A. DELVAUX, T. VELU, M. DE BOER, AND M. GOLDMAN. Interleukin10 inhibits B7 and intercellular adhesion molecule-1 expression on human monocytes. Eur. J. Immunol. 24: 1007–1009, 1994. WILLIAMS, I. R., AND L. L. PERRY. Regulation of transplantation immunity in vivo by monoclonal antibodies recognizing host class II restriction elements. II. Effects of anti-Ia immunotherapy on host T cell responses to graft alloantigens. J. Immunol. 134: 2942–2947, 1985. WILLIAMS, N. S., AND V. H. ENGELHARD. Identification of a population of CD4/ CTL that utilizes a perforin-rather than a Fas liganddependent cytotoxic mechanism. J. Immunol. 156: 153–159, 1996. WOLF, L. A., M. COULOMBE, AND R. G. GILL. Donor antigen-presenting cell-independent rejection of islet xenografts. Transplantation 60: 1164–1170, 1995. WOOD, K., AND D. H. SACHS. Chimerism and transplantation tolerance: cause and effect. Immunol. Today 17: 584–588, 1996. WOODLAND, D. L., AND M. A. BLACKMAN. Retroviral super-antigens and T cells. Int. Rev. Immunol. 8: 311–325, 1992. WU, Y., Y. GUO, A. HUANG, P. ZHENG, AND Y. LIU. CTLA-4-B7 interaction is sufficient to costimulate T cell clonal expansion. J. Exp. Med. 185: 1327–1335, 1997. WU, Y., Y. GUO, AND Y. LIU. A major costimulatory molecule on antigen-presenting cells, CTLA4 ligand A, is distinct from B7. J. Exp. Med. 178: 1789–1793, 1993. YAGITA, H., K. SEINO, N. KAYAGAKI, AND K. OKUMURA. CD95 ligand in graft rejection. Nature 379: 682–682, 1996. YAKURA, H. The role of protein tyrosine phosphatases in lymphocyte activation and differentiation. Crit. Rev. Immunol. 14: 311– 336, 1994. YIN, D., AND C. G. FATHMAN. CD4-positive suppressor cells block allotransplant rejection. J. Immunol. 154: 6339–6345, 1995. YORK, I. A., AND K. L. ROCK. Antigen processing and presentation by the class I major histocompatibility complex. Annu. Rev. Immunol. 14: 369–396, 1996. pra APS-Phys Rev January 1999 INDUCTION OF IMMUNOLOGIC TOLERANCE FOR TRANSPLANTATION 454. ZANDERS, E. D., J. R. LAMB, M. FELDMANN, N. GREEN, AND P. C. BEVERLEY. Tolerance of T-cell clones is associated with membrane antigen changes. Nature 303: 625–627, 1983. 455. ZEEVI, A., M. PAVLICK, R. BANAS, C. BENTLEJEWSKI, K. SPICHTY, A. S. RAO, P. FONTES, A. IYENGAR, R. SHAPIRO, F. DODSON, M. JORDAN, AND S. PHAM. Three years of follow-up of bone marrowaugmented organ transplant recipients: the impact on donor-specific immune modulation. Transplant. Proc. 29: 1205–1206, 1997. 456. ZENG, Y., A. GAGE, A. MONTAG, R. ROTHLEIN, J. R. THISTLETHWAITE, AND J. A. BLUESTONE. Inhibition of transplant rejection by pretreatment of xenogeneic pancreatic islet cells with anti-ICAM-1 antibodies. Transplantation 58: 681–689, 1994. 457. ZHANG, Z. J., L. DAVIDSON, G. EISENBARTH, AND H. L. WEINER. / 9j0d$$ja01 P26-8 141 Suppression of diabetes in nonobese diabetic mice by oral administration of porcine insulin. Proc. Natl. Acad. Sci. USA 88: 10252– 10256, 1991. 458. ZHANG, Z. J., C. S. Y. LEE, O. LIDER, AND H. L. WEINER. Suppression of adjuvant arthritis in Lewis rats by oral administration of type II collagen. J. Immunol. 145: 2489–2493, 1990. 459. ZHENG, L. X., G. FISHER, R. E. MILLER, J. PESCHON, D. H. LYNCH, AND M. J. LENARDO. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature 377: 348–351, 1995. 460. ZHENG, X. X., M. H. SAYEGH, X. G. ZHENG, Y. S. LI, P. S. LINSLEY, R. PEACH, F. BORRIELLO, T. B. STROM, A. H. SHARPE, AND L. A. TURKA. The role of donor and recipient B7–1 (CD80) in allograft rejection. J. Immunol. 159: 1169–1173, 1997. 12-09-98 00:40:31 pra APS-Phys Rev