High molecular weight dissolved organic matter extinction cultures
advertisement

High molecular weight dissolved organic matter
enrichment selects for methylotrophs in dilution to
extinction cultures
The MIT Faculty has made this article openly available. Please share
how this access benefits you. Your story matters.
Citation
Sosa, Oscar A, Scott M Gifford, Daniel J Repeta, and Edward F
DeLong. “High Molecular Weight Dissolved Organic Matter
Enrichment Selects for Methylotrophs in Dilution to Extinction
Cultures.” ISME J 9, no. 12 (May 15, 2015): 2725–2739. © 2015
International Society for Microbial Ecology
As Published
http://dx.doi.org/10.1038/ismej.2015.68
Publisher
Nature Publishing Group
Version
Final published version
Accessed
Thu May 26 17:57:45 EDT 2016
Citable Link
http://hdl.handle.net/1721.1/101411
Terms of Use
Creative Commons Attribution-Noncommercial-Share Alike
Detailed Terms
http://creativecommons.org/licenses/by-nc-sa/4.0/
The ISME Journal (2015) 9, 2725–2739
© 2015 International Society for Microbial Ecology All rights reserved 1751-7362/15
OPEN
www.nature.com/ismej
ORIGINAL ARTICLE
High molecular weight dissolved organic matter
enrichment selects for methylotrophs in dilution to
extinction cultures
Oscar A Sosa1,2,3,5, Scott M Gifford2,5, Daniel J Repeta4 and Edward F DeLong1,2
1
Center for Microbial Oceanography: Research and Education, University of Hawaii, Honolulu, HI, USA;
Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, MA,
USA; 3Biology Department, Woods Hole Oceanographic Institution, Woods Hole, MA, USA and 4Department
of Marine Chemistry and Geochemistry, Woods Hole Oceanographic Institution, Woods Hole, MA, USA
2
The role of bacterioplankton in the cycling of marine dissolved organic matter (DOM) is central to the
carbon and energy balance in the ocean, yet there are few model organisms available to investigate
the genes, metabolic pathways, and biochemical mechanisms involved in the degradation of this
globally important carbon pool. To obtain microbial isolates capable of degrading semi-labile DOM for
growth, we conducted dilution to extinction cultivation experiments using seawater enriched with
high molecular weight (HMW) DOM. In total, 93 isolates were obtained. Amendments using HMW DOM
to increase the dissolved organic carbon concentration 4x (280 μM) or 10x (700 μM) the ocean surface
water concentrations yielded positive growth in 4–6% of replicate dilutions, whereas o1% scored
positive for growth in non-DOM-amended controls. The majority (71%) of isolates displayed a distinct
increase in cell yields when grown in increasing concentrations of HMW DOM. Whole-genome
sequencing was used to screen the culture collection for purity and to determine the phylogenetic
identity of the isolates. Eleven percent of the isolates belonged to the gammaproteobacteria including
Alteromonadales (the SAR92 clade) and Vibrio. Surprisingly, 85% of isolates belonged to the
methylotrophic OM43 clade of betaproteobacteria, bacteria thought to metabolically specialize in
degrading C1 compounds. Growth of these isolates on methanol confirmed their methylotrophic
phenotype. Our results indicate that dilution to extinction cultivation enrichede with natural sources
of organic substrates has a potential to reveal the previously unsuspected relationships between
naturally occurring organic nutrients and the microorganisms that consume them.
The ISME Journal (2015) 9, 2725–2739; doi:10.1038/ismej.2015.68; published online 15 May 2015
Introduction
Marine dissolved organic matter (DOM) supports a
considerable fraction of the carbon, energy and
nutrient requirements of bacterioplankton in the
ocean, yet the biological processes that control
DOM turnover are poorly understood. This is due
in part to the chemical complexity of naturally
occurring DOM and the current level of molecular
characterization, along with a lack of information on
the variety of bacteria and metabolic strategies
involved in DOM degradation.
Marine DOM characterization has been largely
limited to defining its bulk chemical properties.
Marine DOM can be operationally separated into two
Correspondence: EF DeLong, Center for Microbial Oceanography:
Research and Education, University of Hawaii, Honolulu, HI
96822, USA.
E-mail: edelong@hawaii.edu
5
These authors contributed equally to this work.
Received 17 December 2014; revised 4 March 2015; accepted
18 March 2015; published online 15 May 2015
different fractions based on size: low molecular
weight (o1 kDa) and high molecular weight (HMW:
⩾ 1 kDa). The HMW DOM fraction is isolated from
seawater by ultrafiltration and typically accounts for
25–40% of the total dissolved organic carbon (DOC)
in marine waters (Benner, 2002). The elemental
composition of HMW DOM (carbon:nitrogen:phosphorus = 298:18:1; Benner, 2002) contrasts with
marine plankton (Redfield et al., 1963) in that
HMW DOM is depleted in nitrogen and phosphorus
relative to carbon. HMW DOM from open oceansurface waters contains 40–60 nmol nitrogen per
μmol carbon primarily in the form of amides
(McCarthy et al., 1997; Aluwihare et al., 2005) and
only 3–4 nmol phosphorus per μmol carbon as part
of phosphate esters and phosphonate molecules
(Clark et al., 1998; Kolowith et al., 2001). The 13C
NMR spectrum of HMW DOM from marine surface
waters indicates it is largely composed of carbohydrates (50%), humic substances (25%) and a lesser
amount of lipids and proteins (Benner et al., 1992;
Aluwihare et al., 1997; Benner, 2002).
HMW DOM selects for methylotrophs
OA Sosa et al
2726
Radiocarbon analysis offers insight of the fate and
reactivity of DOM. HMW DOM from the North
Pacific has a Δ14C of 10‰ and its constituting neutral
monosaccharides derived from acid hydrolysis have
Δ14C values of 47–67‰. This is similar to the Δ14C of
dissolved inorganic carbon in surface waters
(72 ± 7‰; Repeta and Aluwihare, 2006) and indicates
recent production. In contrast, total DOC is radiocarbon depleted (Δ14C of − 146‰ and − 540‰ in
surface and deep North Pacific waters, respectively)
likely owing to the presence of refractory carbon
(Williams and Druffel, 1987). This data suggests that
subcomponents of HMW DOM from surface waters,
including polysaccharides, cycle faster (1–25 years;
Repeta and Aluwihare, 2006) than the total pool of
DOC (6000 years; Williams and Druffel, 1987). These
chemical characteristics, along with field observations of semi-labile DOM utilization in natural
seawater (Carlson et al., 2004; McCarren et al.,
2010), indicate HMW DOM can fuel microbial
metabolism on ecologically relevant timescales.
Difficulties in identifying the specific chemical
moieties supporting such growth, however, limits
current understanding of DOM transformation,
remineralization and ultimate impact on the flux of
carbon and nutrients through the marine food web.
Bacteria and Archaea are the dominant degraders
of DOM (Carlson, 2002), but the specific taxa, genes
and molecular mechanisms most responsible for
DOM metabolism remain relatively obscure.
Recently, several culture-independent studies have
begun to identify the microbial community members
that respond to DOM enrichment, though the
majority of these studies have focused on DOM
isolated from phytoplankton cultures (Poretsky et al.,
2010; Nelson and Carlson, 2012; Sarmento and
Gasol, 2012; Landa et al., 2013; Sharma and Becker
et al., 2013; Beier et al., 2014), which is presumed to
be more labile than HMW DOM collected in
oligotrophic environments. This is likely because of
the presence of labile proteins and amino acids
(Sarmento et al., 2013) and homopolysaccharides
(Meon and Kirchman, 2001) in phytoplankton DOM.
Aluwihare et al. (1997) and Aluwihare and Repeta
(1999) observed rapid drawdown of phytoplankton
DOM and evolution of polysaccharides from homopolysaccharides (energy storage products) to heteropolysaccharides with a composition like HMW
DOM. In contrast, there have been fewer cultureindependent studies focusing on the cycling of HMW
DOM (McCarren et al., 2010; Sharma and Becker
et al., 2013). In all of these studies, DOM additions
resulted in the enrichment of select members of
the microbial community and in several cases a
temporal succession of DOM-degrading bacteria
(McCarren et al., 2010; Sharma and Becker et al.,
2013). Although these “omic”-based studies have
generated hypothesis as to the functional and
metabolic roles of DOM-degrading bacteria, appropriate model systems upon which to test such
hypothesis and dissect the genes, metabolic
The ISME Journal
pathways and chemical transformation reactions
driving DOM degradation are still lacking.
The goal of this study was to isolate HMW DOM
metabolizing bacteria from marine environments to
develop model systems for investigating the biochemical and metabolic pathways driving DOM transformations in the ocean. Dilution to extinction cultivation
has greatly expanded the collection of marine bacterial
isolates (Button et al., 1993; Connon and Giovannoni,
2002), and has proved instrumental in isolating
organisms resistant to traditional cultivation techniques, including members of the SAR11 and SAR116
clades (Rappé et al., 2002; Stingl et al., 2007a, 2008),
oligotrophic marine gammaproteobacteria (Cho and
Giovannoni, 2004), members of the SUP05 clade of
gammaproteobacterial sulfur oxidizers (Marshall and
Morris, 2013) and pyschropiezophilic alphaproteobacteria (Eloe et al., 2011). Field studies, genome analysis
and pure cultures experiments enabled by the
cultivation of bacterial clades like SAR11 have
expanded our understanding of the ecology and
physiology of marine bacteria and their role in the
ocean carbon cycle (Malmstrom et al., 2004;
Schwalbach et al., 2010; Sun et al., 2011; Grote
et al., 2012). In this study we expanded on the
technique by enriching dilution to extinction cultures
with HMW DOM collected directly from ocean surface
waters, leveraging the high-throughput nature of
dilution to extinction cultivation to screen a microbial
assemblage for HMW DOM-utilizing organisms.
Results
Molecular characterization of HMW DOM
DOM was characterized by 1H nuclear magnetic
resonance spectroscopy to determine major constituents. The spectrum looked similar to previously
published 1H NMR spectra from samples collected at
the Natural Energy Laboratory study site (Repeta and
Aluwihare, 2006) with strong resonances at 5.5 p.p.m.
from carbohydrate anomeric (O-CH-O), 4.0 p.p.m.
from carbohydrate (HCOH), 2.7 p.p.m. from N-acetyl
N-methyl aminosugar (H3C-N-C(O)CH3), 2.0 p.p.m.
from N-acetyl aminosugars N-C(O)CH3) and 1.3 p.p.m.
from 6-deoxysugar protons. The spectral region
between 0.9–3.0 p.p.m. also included a broad unresolved baseline attributed to co-occurring humic
substances. Monosaccharide analysis of hydrolysable
sugars showed the presence of similar amounts of
glucose, galactose, mannose, rhamnose, fucose and
xylose+arabinose. This distribution is typical of
HMW DOM hydrolysable sugars in open ocean
surface seawater (Aluwihare et al., 1997). The sum
of hydrolysable neutral sugars represent 10% of total
HMW DOC.
Experimental overview
The culturing method applied here is a modification
of the 'high-throughput' culturing technique described
HMW DOM selects for methylotrophs
OA Sosa et al
2727
by Connon and Giovannoni (2002) to isolate marine
bacteria in seawater media using dilution to extinction. Our main modification consisted of supplementing filter-sterilized natural seawater with HMW
DOM (41 kDa) isolated by ultrafiltration from
oligotrophic ocean surface waters (Figure 1). To
limit the amount of background DOM in the natural
seawater and to increase the probability of obtaining
isolates responding to the HMW DOM additions we
used relatively nutrient deplete, Sargasso Sea seawater sterilized by tangential-flow filtration (TFF) to
prepare dilution to extinction samples.
The inoculum was collected from Nahant Bay in the
coastal North Atlantic and had an initial cell concentration of 1.5 × 106 (±2.2 × 105 s.d.) cells per ml. The
inoculum was diluted into TFF seawater to a final
concentration of 3 cells per ml and then divided into
three treatments: a no DOM addition (70 μM DOC
background) and two HMW DOM additions (4x DOC
(280 μM) and 10x DOC (700 μM)). One mililiter aliquots
were placed in a 48-well cultivation plates and
incubated at 24 °C in the dark. After 4 weeks of
incubation, the cultures were diluted 10 000-fold into
fresh TFF media containing the appropriate DOM
concentrations to ensure viability before the screening.
Growth screen
After 6–7 weeks of incubation, the cultures were
screened for growth by flow cytometry and any well
with 4105 cells per ml was scored growth positive.
Out of 2520 potential extinction cultures, 93 (3.7%)
HMW
DOM
microbial
TFF
community seawater
scored positive. Only 1 of the 840 non-DOMamended inoculated wells (0.1%) scored growth
positive (Table 1). By contrast, the HMW DOM
amendments had significantly more wells scoring
growth positive (Table 1; Po0.05, bootstrap comparison with 106 iterations), increasing the percent
recovery to 4.7% and 6.0% for the 4x and 10x DOC
treatments, respectively. Cell concentrations also
tended to increase with higher DOM dose
(Figure 2), with upper concentrations reaching
4106 cells per ml (max 2.4 × 106), the majority of
which occurred in the 10x DOC treatment. No
growth was detected in any of the non-inoculated
control wells (Table 1). The single culture obtained
from the non-DOM-amended treatment showed little
sustained growth, and could not be maintained in
successive transfers of the culture collection grown
on 4x DOC.
HMW DOM dose response
To verify that the isolates had a discernible growth
response to the HMW DOM, the 93 cultures were
tested for increased cell yields with DOM additions.
The TFF seawater was first UV-oxidized to reduce
background DOC concentrations from 70 μM DOC to
25 μM DOC. HMW DOM was then added at 4x (280
μM), 20x (1400 μM) or 100x (7000 μM) open ocean
DOC concentrations. Sixty six of the cultures showed
an increase in cell yields with DOM addition,
reaching 45 × 105 cells per ml in the DOMamended treatments and having no growth in the
non-DOM-amended controls (Supplementary Figure
S1). Furthermore, 36 cultures showed a direct
proportional increase in cell yields with increasing
HMW DOM concentrations (100x420x44x).
Dilute to 5-10 cells per ml
Enrich media with DOM
Identification and purity screen
Array one ml aliquots in 48 well plates
In an initial phylogenetic screen of the isolates, 88
small subunit (SSU) rRNA gene sequences were
Incubate
Screen for growth with flowcytometer
Cull to wells with >105 cells ml-1
Transfer to 200
ml media
Incubate
Collect cells
Extract DNA
Nextera XT
library prep
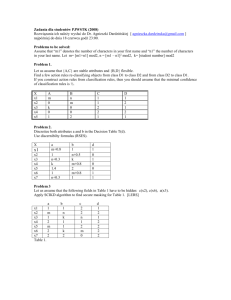
Table 1 HMW DOM-enriched dilution to extinction cultivation
experimental design and growth screen results. Forty-eight well
plates were filled with TFF oligotrophic seawater amended with
HMW DOM at 4x (280 μM) DOC, 10x (700 μM) DOC or nonamended and then inoculated with the diluted coastal
bacterioplankton community
Screen for increased growth
yields on DOM
Regularly transfer to fresh media
and maintain in actively growing
cultures
Wells
Innoculated
Controls
Cryopreserve in DMSO or glycerol
Cultures detected
Innoculated
Controls
Genome
Sequencing
Figure 1 Procedure for the setup, incubation, screening and
downstream analysis of dilution to extinction cultures enriched
with high molecular weight dissolved organic matter (HMW
DOM), including purity screening and phylogenetic identification
via whole-genome sequencing. TFF, tangential-flow filtered.
non-DOM
4 × DOM
10 × DOM
Total
840
54
840
54
840
54
2520
162
1
0
40
0
52
0
93
0
Abbreviation: DOM, dissolved organic matter.
The 'controls' consisted of six wells on each treatment plate, as well as
an entire microtiter plate, that contained the same media and DOM
enrichment, but were not inoculated. Positive growth ('cultures
detected') was defined as a well having a density of 105 or more cells
per ml after 6–7 weeks of incubation and one round of redilution.
The ISME Journal
HMW DOM selects for methylotrophs
OA Sosa et al
2728
40
non-amended
4x DOC
10x DOC
cultures
30
20
10
0
NP
NP
density
(cells ml-1)
Figure 2 Effect of HMW DOM enrichment on dilution to
extinction cultures’ final cell density. Only wells that scored
positive for growth (⩾105 cells per ml) are included. NP, no
positive cultures detected; 4x DOC, ~ 280 μM DOC; 10 × DOC,
~ 700 μM DOC.
determined via PCR amplification and subsequent
Sanger sequencing. Ten of these sequences clustered
within the gammaproteobacteria Alteromonadaceae
(7 cultures including 3 members of the SAR92 clade
and 1 of the OM60 clade), Pseudoalteromonadaceae
(1 culture) and Vibrio (2 cultures). The remaining 78
sequences all clustered within the OM43 clade of
Methylophilaceae betaproteobacteria. There was no
significant difference in the relative recovery of any
specific taxonomic group in the 4x versus 10x DOC
treatments.
The quality of the SSU rRNA Sanger sequencing
provided an initial assessment of culture purity, with
positive PCR amplifications yielding poor quality
Sanger sequences being potentially indicative of
mixed cultures. We applied an additional approach
to assess the purity of cultures and identify their
phylogenetic origins using 'tagmentation' sequencing
library preparation and Illumina paired-end sequencing for whole-genome shotgun (WGS) sequencing
(Figure 1 and see Materials and methods section).
Sixty-eight cultures were successfully sequenced,
with an average of 120 000 paired reads (200 × 200
bases) per sample.
The purity of the cultures was assessed using the
WGS data in three ways. In the first approach, the
reads were de novo assembled into contigs which
were then searched via BLASTn against the SILVA
rRNA database to identify SSU rRNA and large
subunit rRNA sequences. Samples having multiple
rRNA genes binning to different organisms were
considered mixed. Ninety percent of the 68 cultures
had a single copy of small and large subunit rRNA
gene binning to one organism with high identity
(Table 2), either an OM43 clade betaproteobacterium
(82%) or miscellaneous gammaproteobacteria (9%).
Five samples contained multiple rRNA genes hitting
two different organisms. These were primarily an
OM43 clade betaproteobacteria with a SAR92 clade
The ISME Journal
gammaproteobacteria, although one culture (NB0016)
contained an OM43 clade betaproteobacteria and a
flavobacterium.
To verify that rRNAs did not escape detection in the
course of sequence assembly we searched for SSU
rRNA sequences in the unassembled reads. The quality
controlled (trimmed and paired-end joined) unassembled reads were searched against each sample’s
assembled SSU rRNA sequence. Samples having a
large portion of reads matching at low identity were
taken as an indication of a mixed culture. Four of the
five same samples that yielded multiple, divergent
rRNA reads after assembly (Table 2) had high levels
of low identity rRNA matching reads (5–17%;
Supplementary Figures S2 and S3).
Finally, a metagenomic approach was taken, taxonomically binning the unassembled reads for each
sample based on the top hit from a LAST sequence
similarity search against NCBI’s RefSeq database. For
54 of the 60 cultures with betaprotebacterial SSU rRNA
sequences the majority of reads binned to betaproteobacterial reference genomes (Figure 3). The remaining
six cultures had a substantial enrichment in reads
binning to non-betaprotebacterial reference genomes
(17–53% of total reads). Five of these cultures were
also identified as mixed using the SSU rRNA analyses
above but the sixth (NB0010) was only confidently
detected with the metagenomic approach because
the unassembled SSU rRNA analysis revealed only
a marginal proportion of low identity SSU rRNA reads
(1.4%).
In summary, these three screening approaches
produced congruent assessments of culture identity
and purity, with only 6 out of 68 of WGS-sequenced
cultures indicating the presence of mixed populations.
Isolate phylogeny
The WGS-assembled SSU rRNA sequences matched
excellently with the Sanger reads and provided
phylogenetic placement of the culture collection.
Of the 58 WGS SSU rRNA sequences (length = 1542
nucleotides) that had overlapping regions with
quality Sanger sequences (489–1444 bp, quality497%), only five sequences had mismatches
between the Sanger and WGS data (NB0034 (10
mismatches), NB0054 (11), NB0057 (32), NB0068
(12) and NB0072 (194)), with the remaining 53
cultures matching at 100% identity between the
two sequencing methods.
The full-length WGS-assembled rRNA sequences
were used to determine the phylogenetic placement
of cultures. The OM43 clade cultures consisted of 16
unique groups of sequences that clustered with
sequences obtained from coastal metagenomes and
clone libraries (Figure 4). The majority of methylotroph isolates were 499% identical to one another at
the SSU rRNA level with the greatest sequence
divergence occurring between isolate NB0027 and
NB0070 (96.6% SSU rRNA identities, Figure 4).
The gammaproteobacteria sequences were more
1
1
1
1
1
1
1
1
1
1
1
2
2
3
2
2
Md
11
15
30
59
70
72
77
80
93
94
16
48
54
71
76
2
2
2
2
1
1
1
0
1
1
1
1
1
1
1
2
23 S
Methylophilaceae OM43 clade uncultured bacterium
Simiduia agarivorans SA1 DSM 21679
SAR92 clade uncultured bacterium
Algicola bacteriolytica
Vibrio splendidus LGP32
Methylophilaceae OM43 clade uncultured bac. 580
Vibrio splendidus LGP32
Simiduia agarivorans SA1 DSM 21679
Vibrio splendidus LGP32
Methylophilaceae OM43 clade uncultured bac. 580
Alteromonadaceae uncultured bacterium
Methylophilaceae OM43 clade uncultured bacterium
Polaribacter; uncultured Flavobacteriaceae
Methylophilaceae OM43 clade uncultured bac. 580
SAR92 clade uncultured bacterium
Methylophilaceae OM43 clade uncultured bacterium
SAR92 gamma proteobacterium HTCC230
Microbulbifer variabilis Ni-2088
Methylophilaceae OM43 clade uncultured bacterium
Microbulbifer variabilis Ni-2088
Methylophilaceae OM43 clade uncultured bacterium
SAR92 clade uncultured bacterium
Best match
99.4
93.9
98.5
99.1
97.9
99.9
99.8
93.9
99.6
99.9
98.3
99.9
99.8
99.8
98.2
99.3
99.7
92.7
99.7
99.6
99.7
99.6
Percentage IDb
Best matching described taxa via 16 S rRNA
1534
1538
1533
1497
1548
1532
1548
1538
1548
1532
1490
1534
976
1533
1507
1535
758
562
1546
1097
1534
1492
Lengthc
OM43 clade betaproteobacterium KB13
Teredinibacter turnerae T7901
Congribacter; gamma proteobacterium HTCC2207
ND
Vibrionales bacterium SWAT-3
Methylophilaceae OM43 clade uncultured bac. 580
Vibrio splendidus LGP32
Teredinibacter turnerae T7901
Vibrio splendidus LGP32
Methylophilaceae OM43 clade uncultured bac. 580
Glaciecola sp. HTCC2999
Methylophilaceae OM43 clade uncultured bac. 580
Polaribacter irgensii 23-P
Methylophilaceae OM43 clade uncultured bac. 580
Congribacter; gamma proteobacterium HTCC2207
OM43 clade betaproteobacterium KB13
Congribacter; gamma proteobacterium HTCC2207
ND
OM43 clade betaproteobacterium KB13
Congribacter; gamma proteobacterium HTCC2207
OM43 clade betaproteobacterium KB13
Congribacter; gamma proteobacterium HTCC2207
Best match
2874
2624
2909
2902
2883
2879
2871
2905
2883
2882
2874
2402
2883
2903
2874
1348
—
2893
2283
2874
1587
99.9
99.7
99.9
92.5
99.4
99.7
91.9
99.4
95.1
99.7
97.9
98.8
98.2
—
99.7
97.9
99.7
97.9
Lengthc
98.9
92.3
97.9
Percentage IDb
Best matching described taxa via 23 S rRNA
Abbreviations: ND, not detected; bac., bacterium. The number of rRNA genes detected in a given sample was used as an indication of culture purity. The rRNA gene phylogeny was identified by a
BLASTn similarity search against the SILVA SSU and LSU database.
a
Culture identification number (NB00##).
b
Percent matching identities.
c
Gene length.
d
The 53 cultures of 68 cultures screened with a single 16 S and 23 S rRNA gene and which had the top BLASTn match for both genes to the OM43 clade of Methylophilaceae. The mean (± s.d.)
perecent identites for these 53 cultures was 99.4 ( ±0.4) for the 16 S rRNA and 98.9 (±0.05) for the 23 S rRNA.
16 S
IDa
Genes found
Table 2 Phylogenetic identification of HMW DOM-enriched dilution cultures via assembled SSU and LSU rRNA genes from whole-genome sequencing
HMW DOM selects for methylotrophs
OA Sosa et al
2729
The ISME Journal
HMW DOM selects for methylotrophs
OA Sosa et al
2730
Proportion of reads
0
20
40
60
Proportion of reads
80
0
100
20
40
60
80
100
53
54
57
60
61
62
65
66
68
69
70
71
74
75
76
79
81
82
83
84
86
87
88
89
92
93
95
1
3
5
7
8
10
14
16
19
20
21
22
23
25
27
28
29
32
34
35
37
39
40
41
42
45
46
47
48
49
50
51
52
11
15
30
59
72
77
80
94
Betaproteobacteria
Methylophilales
Gammaproteobacteria
SAR92
Alteromonadales
Vibrionales
Alphaproteobacteria
Bacteroidetes
Other
Figure 3 Taxonomic binning of unassembled reads based on the top hit of a LAST sequence similarity search against NCBI’s REFSeq
database. The culture identification number (NB00##) is located on the left of the sample bar.
phylogenetically diverse, spanning several Alteromonadales clades and Vibrio (Supplementary Figure S4).
The SSU rRNA sequence of NB0015 clustered with
the SAR92 clade isolate HTCC2207 (SSU 97%
identity). Four additional SAR92 clade SSU rRNA
sequences were identified as mixed with OM43 clade
cultures and were 98–100% similar to the NB0015
sequence. The remaining Alteromonadales sequences
clustered with Teredinibacter turnerae (NB0011 and
NB0077), Alteromonas macleodii (NB0094) and Psychrosphaera saromensis (NB0030). Vibrio sequences
(NB0059, NB0072 and NB0080) were closely related
to marine strains Vibrio chagasii, Vibrio lentus and
Vibrio pomeroyi (Supplementary Figure S4).
Growth responses to differing media and carbon
substrates
Two isolates representative of the most commonly
identified bacterial groups, OM43 clade betaproteobaThe ISME Journal
cterium NB0046 and SAR92 clade gammaproteobacterium NB0015, were selected for further characterization.
Growth was undetectable for the OM43 betaproteobacterium NB0046 on rich media (Marine Broth
2216), methanol amended agar plates (Janvier et al.
1985) or TFF seawater amended with glucose and
succinate (100 μM each). In non-carbon amended TFF
oligotrophic seawater, NB0046 will typically reach
cell concentrations of 1 × 105 cells per ml, although
we have found this to be variable, with some cultures
showing zero growth in the no carbon amendments,
whereas others can reach almost 106 cells per ml. We
also tested cultures in media prepared with UVoxidized seawater. UV oxidation lowers the total
amount of naturally occurring DOM in the seawater,
but the oxidation process can also produce a broad
suite of low molecular weight, highly oxidized
organic compounds that could serve as substrates or
inhibitors to our isolates (Moran and Zepp, 1997).
However, in our experiments, UV oxidation of the
HMW DOM selects for methylotrophs
OA Sosa et al
2731
Figure 4 Phylogenetic relationships based on the SSU rRNA gene extracted from the whole-genome sequences of Nahant Bay isolates
(NB00##) belonging to the OM43 clade of Betaproteobacteria. The grey box highlights strain NB0046 used in additional growth
experiments (Figure 5). The tree was inferred from 1350 alignment positions from sequences curated in the ARB software (reference) using
the RAxML (maximum likelihood) method. RAxML bootstrap values (1000 replicates) are shown (480%) on nodes. Neighbor-joining
inference method also produced bootstrap values 480% for these nodes. The scale bar indicates substitutions per site.
Gammaproteobacteria and Alphaproteobacteria sequences (not shown) were used as outgroups.
seawater medium consistently reduced growth yields
in non-carbon amended cultures to much lower cell
concentrations (⩽103 cells per ml).
The addition of methanol to seawater media significantly increased OM43 isolate cell yields (Figure 5),
with maximum cell concentrations achieved between
10–100 μM methanol; higher methanol concentrations
(41 mM) tended to inhibit, rather than increase cell
yields, consistent with previous observations of OM43
clade cultures (Halsey et al., 2012). The highest cell
yields were achieved with the HMW DOM additions,
where isolate NB0046 reached 45 × 106 cells per ml in
HMW DOM-containing media at 50x ambient DOC
concentrations (~4 mM DOC, Figure 5). However,
normalizing the observed cell yields to carbon added
to the media indicates that methanol supports higher
growth yields (2.5 × 107 to 2.1 × 108 cells per μmol of
carbon) than HMW DOC treatments (1.6 × 106 to
6.4 × 106 cells per μmol of carbon).
SAR92 clade gammaproteobacterium NB0015 grew
in rich media (Marine Broth 2216), although only at
dilute concentrations (1:10 and 1:100 strength). No
growth was detected on full strength media, marine
agar, seawater amended with methanol, or seawater
amended with glucose and succinate (100 μM each).
In non-carbon amended TFF oligotrophic seawater,
NB0015 reached 1x105 cell per ml. Supplementing
TFF oligotrophic seawater with HMW DOM (4x and
8x DOC) increased NB0015 growth to 2–5 × 105 cells
per ml (Figure 5). The cell yield normalized to
carbon added as HMW DOC was 7.1 × 105 cells per
μmol of carbon, an order of magnitude lower than for
the OM43 clade isolate NB0046. In contrast, cultures
of NB0015 reached on average a maximum cell
concentration of 1.5 × 106 cells per ml when grown in
an artificial seawater medium (Kester et al., 1967)
supplemented with a mix of simple carbon compounds (D-glucose (55 μM), succinate (85 μM), pyruvate
(114 μM), glycerol (109 μM), N-acetyl D-glucosamine
(45 μM) and ethanol (434 μM); Supplementary Figure
S5), consistent with the previous isolation of SAR92
clade strains (Cho and Giovannoni, 2004).
Discussion
Dilution to extinction cultivation has proven a useful
approach for obtaining highly abundant but difficult
The ISME Journal
HMW DOM selects for methylotrophs
OA Sosa et al
2732
Figure 5 Growth profiles of select cultures amended with HMW DOM. Cells from either (a) OM43 clade betaproteobacterium isolate
NB0046 or (b) SAR92 clade gammaprotebacterium isolate NB0015 were inoculated into TFF oligotrophic seawater amended with HMW
DOM and inorganic nutrients (OM43 culture: 400 μM NH4, 30 μM PO4; SAR92 culture: 200 μM NH4, 200 μM NO3, 25 μM PO4). Additional
methanol (MeOH) treatments were included for the OM43 clade isolate. Points are the mean cell concentrations of four (OM43) or three
replicates (SAR92), with error bars representing the s.d. (where not visible, error bars are smaller than the symbols).
to cultivate bacteria. It enables separation of slower
growing cells from faster, more easily cultured
microorganisms, and it also allows the growth of
cells in media that more closely mimics the native
environment. Although several studies have supplemented natural seawater media with defined carbon
substrates in efforts to improve the cell yields (Cho
and Giovannoni, 2004) we applied the alternative
approach of enriching seawater media with HMW
DOM collected directly from the environment.
We postulated that the native, complex mixture of
marine HMW DOM could increase the probability of
obtaining relevant DOM-degrading microorganisms.
Such isolates could then be used as tools to help
define the chemical characteristics of marine HMW
DOM and the biological processes that degrade it.
A similar tactic was successfully applied by HutalleSchmelzer et al. (2010) who enriched bacterioplankton
dilution cultures with the humic fraction of lakederived DOM.
In our study, dilution to extinction cultures
enriched with HMW DOM increased culturing
efficiency and yielded over 90 organisms with
DOM-degrading
capabilities.
Our
non-DOMamended cultures had lower culturing efficiency
(0.1%) than previous dilution culturing studies,
which typically range from 3–25% (Connon and
Giovannoni, 2002; Cho and Giovannoni, 2004; Song
et al., 2009). The reduced culturing efficiency in our
experiments may be explained by the deliberate use
of oligotrophic Sargasso Sea surface water to prepare
the media, which likely has low levels of vitamins
(for example, o0.1 pM vitamin B12 (Menzel and
Spaeth, 1962) and 20–25 pM thiamin (Carini et al.,
2014)), inorganic nutrients (often below detection
limits, 0.05 μmol per kg for nitrate+nitrite and 0.03
μmol per kg for phosphate; http://batsftp.bios.edu/
BATS/bottle) and DOC concentrations (70 μM; as
The ISME Journal
measured for this study) in the basal media than are
typical of coastal seawater. Although this oligotrophic medium may have prohibited the enrichment of some coastal microorganisms, its low DOC
and nutrient background (inorganic nitrogen and
phosphorus were not added) provided greater sensitivity for detecting DOM addition effects without
requiring extensive manipulation of the natural
media. The fact that culturing efficiencies significantly increased in DOM-amended treatments suggested that DOM addition provides, at least partially,
growth factor(s) or organic sources of nitrogen and
phosphorus that were missing in oligotrophic
seawater.
Establishing the purity of the culture collection is
essential for linking the metabolism of DOM to
specific taxa, genes and biochemical mechanisms.
Determining culture purity, however, can be challenging, especially when a culture contains a secondary
isolate at very low abundance (Shrestha et al., 2013).
While SSU rRNA gene sequencing or screens such as
restriction fragment length polymorphisms (RFLP)
are most often used to identify cultures, they suffer
from primer design and preferential amplification
biases which can decrease sensitivity for detecting
mixed cultures. The WGS screen used in this study
does not suffer from oligonucleotide primer mismatch and excludes most other PCR biases thereby
providing a more sensitive and universal method for
determining culture purity. Although PCR-based
SSU rRNA gene sequencing revealed no mixed
cultures in our study, WGS sequencing showed that
6 of the 68 screened cultures were mixed as
congruently determined by three different bioinformatic analyses: (i) number of contigs containing SSU
rRNA genes, (ii) unassembled read’s sequence
similarity to assembled SSU rRNA sequences and
(iii) unassembled read taxon binning based on
HMW DOM selects for methylotrophs
OA Sosa et al
2733
sequence similarity search against NCBI’s RefSeq
database.
The ability of WGS sequencing to identify mixed
cultures is directly dependent on the relative
abundance of the secondary culture in relation to
the sequencing depth, as well as on the phylogenetic
relatedness of the co-isolates. In terms of resolving
mixed cultures of closely related isolates, we found
cultures with no other indication of being mixed had
the majority of unassembled SSU rRNA reads at
⩾ 98% identity to the assembled SSU rRNA sequence
(Supplementary Figure S2), suggesting that two
different populations with o2% SSU rRNA divergence would be difficult to detect. We note that this
would include many of the betaproteobacterial
isolates obtained here. The metagenomic approach
of identifying mixed cultures via taxonomic binning
will be less sensitive than the SSU rRNA bioinformatic approaches to resolving closely related populations owing to database limitations. The breadth of
genes examined using the metagenomic approach,
however, should provide greater sensitivity for
detecting low abundance but phylogenetically
diverse secondary populations. For the betaproteobacterial genomes examined here, 5–10% of
unassembled reads had significant matches to nonbetaproteobacterial genomes likely because of factors
such as short reads lengths, horizontal gene transfer
events and limited reference genomes in the database. This suggests a lower limit for detecting coisolates at around 10% total abundance, though we
note differences in genome size will also affect this
sensitivity. Alternative approaches to increase power
for detecting closely related co-isolates at low
abundance would likely require more sensitive
bioinformatic analysis, such as the statistical frequency of genome single-nucleotide polymorphisms
(Shrestha et al., 2013) focusing on less conserved
genes such as those typically used in multi-locus
sequence typing.
De novo assembly of the WGS data produced
contigs containing full-length SSU and LSU
sequences that were as accurate, and often had
100% identities, with overlapping Sanger reads.
Phylogenetic placement of these WGS SSU rRNA
sequences revealed the culture collection was dominated by members of the OM43 clade of
betaproteobacteria.
The OM43 clade, initially described by Rappé
et al. (1997), was first isolated from the Oregon coast
by Connon and Giovannoni (2002). Their distribution is generally limited to the coastal zone, where
they can compose 1–3% of the bacterioplankton
community (Suzuki et al., 2004), though their
abundance has been shown to significantly increase
during phytoplankton blooms (Morris et al., 2006;
Rich et al., 2011). Metatranscriptomic and metaproteomic data suggest they are active components of
coastal bacterioplankton communities (Sowell et al.,
2010; Gifford et al., 2014). The OM43 isolates
obtained in this study were closely related to those
previously characterized coastal strains and
sequences (Figure 4). The majority of our isolates
clustered most closely with the Hawaiian strain
HIMB624 (Huggett et al., 2012) and related SSU
rRNA phylotypes identified in diverse coastal locations. Three of our isolates formed an outgroup with
the Oregon coastal isolate HTCC2181, as well as
sequences obtained primarily from higher latitudes.
Though closely related, the presence of SSU rRNA
microdiversity within our isolate collection suggests
the prevalence of OM43 clade members in the
dilution to extinction cultures was not because of
the enrichment of a single clonal population.
Genome sequences of OM43 betaproteobacteria
are missing the E1 subunit of the α-ketoglutarate
dehydrogenase complex of the TCA cycle, a trait
thought to be indicative of an obligatory methylotrophic lifestyle (Halsey et al., 2012; Huggett et al.,
2012). A preliminary genome analysis of NB0046
shows that it is also missing the E1 α-ketoglutarate
dehydrogenase subunit. Furthermore, NB0046
shares similar characteristics with other OM43 lineage genomes (Giovannoni et al., 2008; Huggett et al.,
2012), including a small genome size (o1400 genes),
the presence of an alternative methanol dehydrogenase (XoxF) instead of the canonical Mxa or Mdh
methanol dehydrogenase (Chistoserdova, 2011),
genes for formaldehyde oxidation via tetrahydrofolate but not via tetrahydromethanopterin, and
carbon assimilation through the RUMP cycle instead
of the serine cycle. These observations suggest the
methylotrophs isolated on HMW DOM have a
similar 'obligate' methylotrophic lifestyle as other
OM43 clade members, although a more detailed and
thorough analysis of the 60 OM43 genomes
sequenced here will be required to confirm this.
Given their reliance on single carbon substrates,
the predominance of C1 compound specialists in our
cultures amended with HMW DOM was unexpected.
Our results, however, are predicated in a previous
study by McCarren et al. (2010), who observed that
HMW DOM added to oligotrophic ocean bacterioplankton microcosms increased the relative abundance and transcriptional activity of methylotrophs,
particularly Methylophaga species belonging to the
gammaproteobacteria. McCarren et al. (2010) postulated that HMW DOM polysaccharides containing a
large fraction of methylated sugars (Quan and
Repeta, 2007) were the source of single carbon
compounds, including methanol, which provided
the methylotrophs their growth substrate. We further
propose that the prevalence of pure cultures of
methylotrophs in our HMW DOM-enriched dilution
to extinction samples is owing to their ability to
cleave C1 groups from the methyl sugars and uronic
acid methyl esters in the DOM polysaccharide. In
addition, low levels of abiotic oxidation of the HMW
DOM during collection and storage may possibly
degrade the methylated sugars, releasing low molecular weight compounds that can be directly
consumed by methylotrophs. In either case, the data
The ISME Journal
HMW DOM selects for methylotrophs
OA Sosa et al
2734
indicated that HMW DOM can serve as a sole source
of carbon and energy for these methylotrophs.
The order of magnitude difference in growth yields
(cell yield normalized to carbon added) between
methanol (6.8 × 107 to 5.3x108 cells per μmol of
carbon) and HMW DOM (1.6 × 106 to 6.4 × 106 cells
per μmol of carbon) suggests that growth on HMW
DOM is less efficient than on methanol. Similar low
growth yields (7.1 × 105 cells per μmol of carbon) were
observed for SAR92 clade strain NB0015 in cultures
amended with HMW DOM. Alternatively, the low
growth yields observed may indicate that only a small
fraction of HMW DOM may be available to a single
organism with limited metabolic potential, like isolates NB0046 and NB0015, despite the total amount of
DOC added to cultures. The efficiency at which DOC
is converted into biomass, typically o20% in the
open ocean (Carlson and Ducklow, 1996) has implications on the magnitude of carbon that cycles through
bacterioplankton. Measurements of DOC drawdown
and respiration, though not obtained in HMW DOM
dose-response experiments, will be necessary to
determine bacterial growth efficiencies on HMW
DOC and to estimate the bacterial carbon demand
that this organic carbon pool can support.
The second major taxonomic group found in the
HMW DOM-enriched cultures was the gammaproteobacteria. This included isolates belonging to the Alteromonadales and Vibrionales. The Vibrio (NB0059,
NB0072 and NB0080), Alteromonad (NB0094) and
Pseudoalteromonad (NB0030) isolates did not exhibit
a proportional increase in cell yields in response to
added HMW DOM in dose-response experiments even
though these isolates were initially obtained from the
HMW DOM-enriched dilution to extinction cultures.
Among the gammaproteobacteria only strains NB0011
and NB0077, and the SAR92 clade isolates exhibited
increased growth in the presence of HMW DOM. The
SAR92 clade isolate NB0015 exhibited the strongest
growth response to HMW DOM although cell yields
were low (2–4 × 105 cells per ml) compared with the
OM43 clade isolates (45 × 106 cells per ml). However,
NB0015 growth yields reached 41x106 cells per ml
when cultured in defined media with a mixture of
simple carbon substrates indicating that this isolate may
require a variety of carbon compounds and nutrients
not available in HMW DOM to supplement its
metabolism. The SAR92 clade was the most frequently
recovered gammaproteobacteria group in the dilution to
extinction cultures including four SAR92 clade-related
SSU rRNA sequences found mixed with cultures of
OM43 clade strains, more than any other taxa. Including
these additional sequences, the SAR92 clade was the
second most common group identified in our dilution
to extinction cultivation experiment.
The growth of SAR92 clade strains in our HMW
DOM-amended samples may partially be explained
by its numerical abundance in coastal bacterioplankton assemblages (Stingl et al., 2007b) and their ease
of recovery by dilution to extinction cultivation
(Cho and Giovannoni, 2004). These factors alone,
The ISME Journal
however, do not solely account for the prevalence of
SAR92 in our cultures because SAR92 was only
identified in the HMW DOM-enriched dilution to
extinction cultures and not in the non-DOMamended cultures. We postulate that the
carbohydrate-rich component of HMW DOM in
particular may have stimulated the growth of
SAR92 bacteria. Some of the closest relatives of the
SAR92 clade (90–93% SSU rRNA identity) include
cultured isolates with carbohydrate degrading capabilities. For example, Microbulbifer hydrolyticus
(Gonzalez et al., 1997), Saccharophagus degradans
2–40 (Andrykovitch and Marx, 1988; Weiner et al.,
2008) and Simiduia agarivorans SA1 (Shieh et al.,
2008) can breakdown several recalcitrant polysaccharides, including agar, alginate, cellulose or chitin.
Another relative, Teredinibacter turnerae T7902,
a bacterium associated with wood-boring bivalves,
is capable of digesting cellulose (Distel et al., 2002).
It is plausible that SAR92 clade strains may directly
degrade HMW DOM polysaccharides in contrast to
the OM43 clade methylotrophs which may utilize
C1 compounds that decorate the polysaccharides.
The availability of model laboratory organisms able
to grow on HMW DOM now allows us to test these
hypotheses. Future work chemically characterizing
the HMW DOM before and after microbial degradation, as well as examining the transcriptional and
proteomic responses of the isolates during DOM
metabolism will help to determine the bonded
nature of the carbon sustaining the cultures.
In summary, dilution to extinction cultures
enriched with HMW DOM extended the power of
the dilution cultivation technique by enriching for
cells in media closely mimicking their native
environment, and also by stimulating growth using
naturally derived organic substrates. This approach is
useful for obtaining model DOM-degrading isolates as
both the carbon substrates and the organisms acting
upon them are unknown. Using this technique we
found organisms ranging from obligate methylotrophs
to less fastidious heterotrophs that were able to grow
using oligotrophic ocean HMW DOM as a substrate,
suggesting there are multiple metabolic strategies
involved in the degradation of HMW DOM. We note,
however, that only a fraction of the total DOM added
to our cultures was remineralized, suggesting that
there were other growth-limiting factors, or potential
requirement for syntrophic microbial partners to
further degrade the DOM polymers (McCarren et al.,
2010). Co-culture experiments will likely be essential
to further elaborate the biological, physiological and
biochemical details of consortial DOM degradation
processes in the sea.
Materials and methods
Concentration of HMW DOM from seawater
DOM collection was conducted using the method
described by Repeta and Aluwihare (2006). Briefly,
HMW DOM selects for methylotrophs
OA Sosa et al
2735
the HMW fraction of DOM (41 kDa) was concentrated from 24 000 l of surface seawater (15 m)
collected 2 km offshore of the Island of Hawaii,
Hawaii at the National Energy Laboratory Hawaii
Authority (NELHA) in February 2006. The HMW
DOM-concentrated seawater was then filtered
through a 30 kDa ultrafiltration membrane to remove
cell debris and viral particles, diafiltered to remove
salts and freeze-dried. A total of 9.5 g of freeze-dried
HMW DOM was obtained that was 32% carbon,
2.7% nitrogen with a C/N ratio of 13.9 representing
18% of the DOC in the original raw seawater.
filtered through a 142-mm diameter, 0.2-μm poresize Supor-membrane (Pall, Ann Arbor, MI, USA)
and a 0.1-μm pore-size Supor capsule filter (Pall) and
collected in an acid-cleaned polycarbonate carboy.
Pre-filtered water circulated through a Pellicon 2
Mini tangential-flow ultrafiltration system (Millipore, Billerica, MA, USA) consisting of a 30 kDa
cassette of regenerated cellulose (Millipore). The
tangential-flow filtrate was collected in autoclaved,
acid-cleaned polycarbonate carboys and stored at 4°C.
Seawater remained sterile at room temperature after
the addition of nutrients and sterile rich media. The
final TFF seawater medium had a total organic
carbon concentration of 70 μM.
Molecular characterization of HMW DOM
DOM was characterized by 1H nuclear magnetic
resonance spectroscopy to determine major constituents. Spectra were recorded on a Bruker Avance
DPX 400 MHz spectrometer fitted with an inverse
broadband 5 mm probe. Approximately 1–2 mg of
sample was dissolved in 100% D2O. Chemical shifts
were referenced to residual HOD at δ = 4.80 p.p.m.
Acid hydrolysis (2.8 M trifluoroacetic acid, heated at
120°C for 4 hours under nitrogen) of HMW DOM
yields a suite of neutral sugars (arabinose, fucose,
galactose, glucose, mannose, rhamnose, xylose) that
were separated by reverse-phase high-pressure
liquid chromatography (Ascentis C-18 column,
Sigma-Aldrich, St Louis, MO, USA; 150x1 mm,
3 μm, eluted at 120 μl min − 1 with 10/90 (v/v)
acetonitrile/water) and quantified at 307 nm as the
their aminobenzoate ethyl ester derivatives
(Baik and Cheong, 2007). Our chromatographic
analysis does not separate xylose and arabinose
which are reported as the sum xylose+arabinose.
Analyses of unhyrdolyzed HMW DOM does not
yield any aminobenzoate ethyl ester products that
interfere with our monosaccharide analyses.
DOC measurements
Samples for DOC concentration analysis were transferred into combusted glass vials and acidified with
25% phosphoric acid solution before sealing with
acid-washed Teflon septa. Sample concentrations
were determined using the high-temperature combustion method on a Shimadzu TOC-VCSH with
platinized alumina catalyst alongside potassium
hydrogen phthalate standards and consensus reference materials provided by the DOC-CRM program.
Preparation of seawater media
The water for extinction culturing and subsequent
cultivation media was collected off Bermuda
(33.2497°N 65.7103°W) during the KN207-01 cruise,
aboard the R/V Knorr on 03 May 2012 via the ship’s
flow-through system with a 0.2 μm filter. After
collection and storage, the Sargasso seawater was
sterilized by TFF as described by Becker et al. (2007),
with minor modifications. The seawater was pre-
Extinction culturing with HMW DOM additions
The inoculum was collected in Nahant Bay, MA
(42°25.195 N, 70°54.463 W) on 23 August 2012.
Bacterioplankton cell densities were obtained by
direct-cell counts of DNA-stained cells using SYBR
green I (Molecular Probes, Life Technologies, Carlsbad, CA, USA) ~ 1 hour after sample collection. The
inoculum was diluted into sterile seawater to an
estimated cell density of 3 cells per ml in a final
volume of 5 l. The dilute inoculum was split into
three 1-l aliquots, one left untreated (70 μM DOC) and
two were supplemented with HMW DOM to 210 and
630 μM HMW DOC to obtain media with a final 280
(4x) and 700 (10x) μM DOC, respectively. One-ml
aliquots from each treatment were then distributed
into non-tissue culture treated polystyrene 48-well
plates (BD Biosciences, Franklin Lakes, NJ, USA).
Each experimental treatment consisted of 840 potential extinction cultures in twenty 48-well plates.
Every plate included six negative controls consisting
of wells filled with the corresponding treatment
media made up with sterile seawater. Plates were
covered in foil to reduce evaporation and incubated
at 24 °C in the dark for 4–5 weeks. In order to ensure
that potential extinction cultures remained viable
and capable of growth in the corresponding cultivation media, after the 4–5-week incubation the
samples were re-diluted 10 000-fold into new plates
containing fresh TFF seawater and the appropriate
DOM concentrations. These were then incubated for
6–7 weeks under the same conditions as before.
Detection of culture growth
Extinction cultures were detected using a guava
easyCyte 8HT flow cytometry system (Millipore) by
transferring 50 μl from each cultivation plate well to
a 96-well microtitter plate containing 10 μl of 500fold diluted SYBR green I and 140 μl of sterile
seawater. Samples were stained for at least 30 min
before flow cytometry. Each well was analyzed for
15 s at a flow rate of 0.59 μl s − 1 with a blue laser
(488 nm excitation) to detect green fluorescence.
Wells were scored positive for cell growth when
the concentration was ⩾ 105 cells per ml.
The ISME Journal
HMW DOM selects for methylotrophs
OA Sosa et al
2736
HMW DOM dose response screen of the culture
collection
Cultures scoring positive for growth were examined
for increased cell yields under different HMW DOM
concentrations. To limit cell growth to consumption
of HMW DOM added, background DOC in the TFF
seawater was photo-oxidized via exposure to highintensity UV light for 4 h, reducing DOC concentrations by 30–40%. During this process, approximately
an eighth of the volume was lost owing to evaporation, and this was replenished with ultrapure water.
The UV-oxidized TFF seawater (25 μM DOC) was
then supplemented with 210, 1330 and 6930 μM DOC
using HMW DOM to obtain ~ 4x, ~ 20x or ~ 100x
DOC media, respectively, in similitude to media
prepared for extinction culturing. Non DOMamended UV-oxidized seawater served as control.
No inorganic nutrients or vitamins were added to the
media. Each treatment media was distributed into
1-ml aliquots in 48-well cultivation plates. Wells
were inoculated with 10-μl sub-samples of the
dilution to extinction cultures and incubated at 24°C
in the dark. Subsamples were taken every 5–7 days
for cell-density enumeration using the SYBR green I
flow cytometry assay described above.
SSU rRNA sequencing
A total of 400–800 μl from the 93 cultures scoring
positive for growth were transferred to a 0.2 μm
Supor-membrane 96-well filter plate (Pall) and
vacuum filtered. The cells were resuspended from
the filter with two separate aliquots of 125 μl of lysis
buffer (40 mM EDTA, 50 mM Tris at pH 8.3, 0.73 M
sucrose 1.15 mg ml − 1 lysozyme (Sigma-Aldrich),
200 μg ml − 1 RNase (Qiagen, Hilden, Germany) and
transferred to a 96 deep-well plate, which was then
placed at − 80°C. After freezing, the plate was
thawed, Proteinase K and SDS were added to final
concentrations of 0.65 mg ml − 1 and 10% SDS,
respectively, and allowed to incubate for 2 h at 55°C.
The lysate was then transferred to new 2-ml deep-well
plate and DNA extracted using a DNeasy 96-well
Blood and Tissue kit (Qiagen).
The SSU rRNA gene was PCR amplified using
universal Eubacterial primers pA Escherichia coli
8-28 F (5′-AGA GTT TGA TCC TGG CTC AG-3′) and
E. coli 1510-1492R (5′- GGT TAC CTT GTT ACG
ACT T -3’) in 50-μl reactions consisting of 1 μM each
forward and reverse primer, 1 μl of FailSafe Enzyme
Mix (Epicentre, Madison, WI, USA), 25 μl of 2x
FailSafe PCR PreMix E (Epicentre) water and 20-μl
DNA template. After amplification, the PCR amplicons were run on 1% agarose gel and bands running
at ~ 1500–1600 nt were excised and PCR-purified
with a 96-well gel extraction kit (Qiagen) followed by
an additional purification using the QIAquick 96well PCR purification kit (Qiagen) and eluted in 80 μl
elution buffer. Sanger sequencing was conducted
following the BigDye v3.1 sequencing protocol
(Applied Biosystems, Foster City, CA, USA) on a
The ISME Journal
ABI 3730 DNA analyzer (Applied Biosystems) using
the SSU rRNA universal primers described above
(pA E. coli 8-28, E. coli 1510-1492) and with bacterial
primers 519F 5′-CAGCMGCCGCGGTAATWC-3′ and
800R 5′-TACCAGGGTATCTAATCC-3′.
Whole-genome sequencing
Seventy cultures were selected for whole-genome
sequencing based on their growth yields and
response to HMW DOM substrate addition. To obtain
sufficient amounts of DNA for the NexterXT library
preparation, 10-μl of culture was inoculated into
acid-cleaned polycarbonate bottles containing
200 ml TFF seawater amended with HMW DOM to
3x DOC. After incubating at 22°C in the dark for
4–5 weeks, the cells were collected on 0.1 μm poresize, 25 mm diameter Durapore membranes (Millipore) using Swinnex filter-holders (Millipore) and
peristaltic pumping. Filters were stored frozen (−80°C)
in 600 μl of tissue lysis buffer (Qiagen). DNA was
extracted from thawed filters using a DNeasy Blood
and Tissue Kit (Qiagen) following the manufacturer's
instructions. DNA samples were prepared for
sequencing and barcoded using the Nextera XT
DNA 96-sample preparation kit (Illumina, San Diego,
CA, USA), and sequenced with one 250 × 250 nt
paired-end MiSeq run (Illumina). Sequences are
deposited in the NCBI sequence read archive under
study SRP045600.
Bioinformatic and Phylogenetic analysis
FastQ files from the MiSeq run were imported into
the CLC Genomics Workbench (CLC bio, Aarhus,
Denmark). Paired reads were joined and then
assembled into contigs using CLC’s de novo assembler with automatic word and bubble sizes, a
minimum contig length of 200, insertion and
deletion costs set to 3, mismatch cost set to 2, length
fraction set to 0.5 and the similarity fraction set
to 0.8. Only contigs ⩾ 1000 nt in length were further
examined. Contigs containing small and large
subunit rRNA sequences were identified and annotated by a BLASTn sequence similarity search
against the SILVA database (http://www.arb-silva.
de, version 111).
To ensure that low abundance reads with
a divergent SSU rRNA were not missed during the
assembly process the unprocessed miSeq reads were
trimmed and paired-end joined using trimomatic
(http://www.usadellab.org/cms/?page=trimmomatic)
and PandaSeq (https://github.com/neufeld/pandaseq/
wiki/PANDAseq-Assembler), respectively. The trimmed
and joined reads were compared with the assembled
SSU rRNA for each sample via a BLASTn sequence
similarity search, with hits having an e-valueo0.001,
a read length450 nt and 45% identity over the BLAST
alignment considered significant. The unassembled, but
trimmed and joined reads, were also taxonomically
binned based on a LAST (Frith et al., 2010; Kielbasa
HMW DOM selects for methylotrophs
OA Sosa et al
2737
et al., 2011) sequence similarity search against NCBI’s
REfSeq database (version 64) with a score penalty for
frameshift of 500 and an initial match multiplicity of 10.
Top hits with a score ⩾ 40 and alignment length ⩾ 75 nt
were considered significant and used for the taxonomic
annotation.
Partial SSU rRNA gene sequences obtained via
Sanger sequencing were quality-trimmed, manually
inspected and assembled with Sequencher version
5.1 (GeneCodes, Ann Arbor, MI, USA). SSU rRNA
gene sequences from Sanger or WGS were aligned
using the SILVA incremental aligner online tool
(Pruesse et al., 2012) and curated in ARB (Ludwig
et al., 2004) using the All-Species Living Tree project
release 115 (Munoz and Yarza et al., 2011). Fulllength SSU rRNA gene sequences obtained
from WGS were used to build phylogenetic trees
and were deposited in NCBI GenBank under
accession numbers KP770034 through KP770106.
For the betaproteobacteria OM43 clade strains a
tree was inferred from 1350 alignment positions
using the RAxML maximum likelihood method
(Stamatakis, 2014) and neighbor joining (Saitou and
Nei, 1987). For gammaproteobacteria isolates, a tree
was constructed using 1293 unambiguous alignment
positions using RAxML. RAxML 8.0.24 was implemented on Cipres Science Gateway (Miller et al.,
2010) and neighbor joining was implemented on
MEGA5 (Tamura et al., 2004, 2011). RAxML trees
were curated in EMBL’s interactive Tree of Life tool
(Letunic and Bork, 2006, 2011).
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
We thank Jamie Becker for DOC concentration analyses,
technical assistance and providing advice and feedback. We
are also indebted to Tsultrim Palden for assistance in DNA
extraction and sequencing. This research was funded by the
Gordon and Betty Moore Foundation through Grant
GBMF3298 to DJR and EFD, GBMF #3777 (to EFD) and
NSF Science and Technology Center grant EF0424599 (to
EFD), and the Simons Foundation (to EFD and DR). This
work is a contribution of the Center for Microbial Oceanography: Research and Education (C-MORE) and the Simons
Collaboration on Ocean Processes and Ecology (SCOPE).
References
Aluwihare LI, Repeta DJ, Chen RF. (1997). A major
biopolymeric comoponent of dissolved organic carbon
in surface sea water. Nature 387: 166–169.
Aluwihare LI, Repeta DJ. (1999). A comparison of the
chemical characteristics of oceanic DOM and extracellular DOM produced by marine algae. Mar Ecol Prog
Ser 186: 105–117.
Aluwihare LI, Repeta DJ, Pantoja S, Johnson CG. (2005).
Two chemically distinct pools of organic nitrogen
accumulate in the ocean. Science 308: 1007–1010.
Andrykovitch G, Marx I. (1988). Isolation of a new
polysaccharide-digesting bacterium from a salt marsh.
Appl Environ Microbiol 54: 1061–1062.
Baik YS, Cheong WJ. (2007). Determination of molecular
weight distribution and average molecular weights of
oligosaccharides by HPLC with a common C18 phase
and a mobile phase with high water content. Bull
Korean Chem Soc 28: 847–850.
Becker JW, Brandon ML, Rappé MS, Hurst CJ, Crawford
RL, Garland JL et al. (2007). Cultivating microorganisms from dilute aquatic environments: melding traditional methodology with new cultivation techniques
and molecular methods. Manual of Environmental
Microbiology 3: 399–406.
Beier S, Rivers AR, Moran MA, Obernosterer I. (2014). The
transcriptional response of prokaryotes to phytoplanktonderived dissolved organic matter in seawater. Environ
microbiole; doi:10.1111/1462-2920.12434.
Benner R, Pakulski JD, McCarth M, Hedges JI, Hatcher PG.
(1992). Bulk chemical characteristics of dissolved
organic matter in the ocean. Science 255: 1561–1564.
Benner R. (2002). Chemical composition and reactivity. In:
Hansel DA, Carlson CA (eds), Biogeochemistry of
marine dissolved organic matter. Academic Press:
San Diego, CA, USA, pp 59–90.
Button DK, Schut F, Quang P, Martin R, Robertson BR.
(1993). Viability and isolation of marine bacteria by
dilution culture: theory, procedures, and initial results.
Appl Environl Microbiol 59: 881–891.
Carini P, Campbell EO, Morré J, Sañudo-Wilhelmy SA,
Thrash JC, Bennet SE et al. (2014). Discovery of a
SAR11 growth requirement for thiamin’s pyrimidine
precursor and its distribution in the Sargasso Sea.
ISME J 8: 1727–1738.
Carlson CA. (2002). Production and Removal Processes. In:
Hansel DA, Carlson CA (eds), Biogeochemistry of
marine dissolved organic matter. Academic Press:
San Diego, USA, pp 91–151.
Carlson CA, Ducklow HW. (1996). Growth of bacterioplankton and consumption of dissolved organic carbon
in the Sargasso Sea. Aquat Microb Ecol 10: 69–85.
Carlson CA, Giovannoni SJ, Hansell DA, Goldberg SJ,
Parsons R, Vergin K. (2004). Interactions among
dissolved organic carbon, microbial processes, and
community structure in the mesopelagic zone of the
northwestern Sargasso Sea. Limnol Oceanogr 49:
1073–1083.
Chistoserdova L. (2011). Modularity of methylotrophy,
revisited. Environ Microbiol 13: 2603–2622.
Cho J, Giovannoni SJ. (2004). Cultivation and growth
characteristics of a diverse group of oligotrophic
marine Gammaproteobacteria. Appl Environ Microbiol
70: 432–440.
Clark LL, Ingall ED, Benner R. (1998). Marine phosphorus
is selectively remineralized. Nature 393: 426.
Connon S, Giovannoni SJ. (2002). High-throughput methods for culturing microorganisms in very-low-nutrient
media yield diverse new marine isolates. Appl Environ
Microbiol 68: 3878–3885.
Distel DL, Morrill W, MacLaren-Toussaint N, Franks D,
Waterbury J. (2002). Teredinibacter turnerae gen. nov.,
sp. nov., a dinitrogen-fixing, cellulolytic, endosymbiotic gamma-proteobacterium isolated from the gills of
The ISME Journal
HMW DOM selects for methylotrophs
OA Sosa et al
2738
wood-boring molluscs (Bivalvia: Teredinidae). Int J
Syst Evol Microbiol 52: 2261–2269.
Eloe EA, Malfatti F, Gutierrez J, Hardy K, Schmidt WE,
Pogliano K et al. (2011). Isolation and characterization
of a psychropiezophilic alphaproteobacterium. Appl
Environ Microbiol 77: 8145–8153.
Frith MC, Wan R, Horton P. (2010). Incorporating sequence
quality data into alignment improves DNA read
mapping. Nucleic Acids Res 38: e100.
Gifford SM, Sharma S, Moran MA. (2014). Linking
activity and function to ecosystem dynamics in a
coastal bacterioplankton community. Front Microbiol
5: 185.
Giovannoni SJ, Hayakawa DH, Tripp HJ, Stingl U, Givan SA,
Cho JC et al. (2008). The small genome of an abundant
coastal ocean methylotroph. Env Microb 10: 1771–1782.
Gonzalez JM, Mayer F, Moran MA, Hodson RE, Whitman
WB. (1997). Microbulbifer hydrolyticus gen. nov., sp.,
nov., and Marinobacterium georgiense gen. nov., sp.
nov., two marine bacteria from a lignin-rich pulp mill
waste enrichment community. Int J Syst Bacteriol 47:
369–376.
Grote J, Thrash C, Huggett MJ, Landry Z, Carini P,
Giovannoni S et al. (2012). Streamlining and core
genome conservation among highly divergent members of the SAR11 clade. mBio 3: e00252–12.
Halsey KH, Carter AE, Giovannoni SJ. (2012). Synergistic
metabolism of a broad range of C1 compounds in the
marine methylotrophic bacterium HTCC2181. Environ
Microbiol 14: 630–640.
Huggett MJ, Hayakawa DH, Rappé MS. (2012). Genome
sequence of strain HIMB624, a cultured representative
from the OM43 clade of marine Betaproteobacteria.
Stand Genomic Sci 6: 11.
Hutalle-Schmelzer KML, Zwirnmann E, Krüger A,
Grossart HP. (2010). Enrichment and cultivation of
pelagic bacteria from a humic lake using phenol and
humic matter additions. FEMS Microbiol Ecol 72:
58–73.
Janvier M, Frehel C, Grimont F, Gasser F. (1985).
Methylophaga marina gen. nov., sp. nov. and Methylophaga thalassica sp. nov., marine methylotrophs. Int
J Syst Bacteriol 35: 131–139.
Kester DR, Duedall IW, Connors DN, Pytkowicz RM.
(1967). Preparation of artificial seawater. Limnol
Oceanogr 12: 176–179.
Kielbasa SM, Wan R, Sato K, Horton P, Frith MC. (2011).
Adaptive seeds tame genomic sequence comparison.
Genome Res 21: 487.
Kolowith LC, Ingall ED, Benner R. (2001). Composition and
cycling of marine organic phosphorus. Limnol Oceanogr 46: 309–320.
Landa M, Cottrell MT, Kirchman DL, Kaiser K, Medeiros PM,
Tremblay L et al. (2013). Phylogenetic and structural
response of heterotrophic bacteria to dissolved organic
matter of different chemical composition in a continuous
culture study.Environ Microbiol 16: 1668–1681.
Letunic I, Bork P. (2006). Interactive Tree Of Life (iTOL): an
online tool for phylogenetic tree display and annotation. Bioinformatics 23: 127–128.
Letunic I, Bork P. (2011). Interactive Tree of Life v2: online
annotation and display of phylogenetic trees
made easy. Nucleic Acids Res 39: W475–W478.
Ludwig W, Strunk O, Westram R, Ritcher L, Meier H,
Yadhukumar et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32:
1363–1371.
The ISME Journal
Malmstrom RR, Kiene RP, Cottrell MT, Kirchman DL.
(2004). Contribution of SAR11 bacteria to dissolved
dimethylsulfoniopropionate and amino acid uptake in
the North Atlantic ocean. Appl Environ Micorbiol 70:
23–37.
Marshall T, Morris RM. (2013). Isolation of an aerobic
sulfur oxidizer from the SUP05/Arctic96BD-19 clade.
ISME J 7: 452–455.
McCarren J, Becker JW, Repeta DJ, Shi Y, Young CR,
Malmstrom RR et al. (2010). Microbial community
transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc Natl Acad Sci USA 107:
16420–16427.
McCarthy M, Pratum T, Hedges J, Benner R. (1997).
Chemical composition of dissolved organic nitrogen
in the ocean. Nature 390: 150–154.
Menzel DW, Spaeth JP (1962). Occurrence of vitamin B12
in the Sargasso Sea. Limnol Oceongr 7: 151–158.
Meon B, Kirchman DL. (2001). Dynamics and molecular
composition of dissolved organic material during
experimental phytoplankton blooms. Mar Chem 75:
185–199.
Miller MA, Pfeiffer W, Schwartz T. (2010). Creating the
CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing
Environments Workshop (GCE), 14 November 2010,
New Orleans, LA, USA, pp 1–8.
Moran MA, Zepp RG. (1997). Invited ReviewRole of
photoreactions in the formation of biologically labile
compounds from dissolved organic matter. Limonol
Oceanogr 42: 1307–1316.
Morris RM, Longnecker K, Giovannoni SJ. (2006). Pirellula
and OM43 are among the dominant lineages identified
in an Oregon coast diatom bloom. Environ Microbiol 8:
1361–1370.
Munoz R, Yarza P, Ludwig W, Euzéby J, Amann R,
Schleifer KH et al. (2011). Release LTPs104 of the
all-species living tree. Syst Appl Microbiol 34:
169–170.
Nelson CE, Carlson CA. (2012). Tracking differential
incorporation of dissolved organic carbon types among
diverse lineages of Sargasso Sea bacterioplankton.
Environ Microbiol 14: 1500–1516.
Poretsky RS, Sun S, Mou X, Moran MA. (2010). Transporter genes expressed by coastal bacterioplankton in
response to dissolved organic carbon. Environ Microbiol 12: 616–627.
Pruesse E, Peplies J, Glöckner FO. (2012)SINA: accurate
high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28: 1823–1829.
Quan TM, Repeta DJ. (2007). Periodate oxidation of marine
high molecular weight dissolved organic matter:
evidence for a major contribution from 6-deoxy-and
methyl sugars. Mar Chem 105: 183–193.
Rappé M, Kemp PF, Giovannoni SJ. (1997). Phylogenetic
diversity of marine coastal picoplankton 16S rRNA
genes cloned from the continental shelf off Cape
Hatteras, North Carolina. Limnol Oceanogr 42:
811–826.
Rappé MS, Connon SA, Vergin KL, Giovannoni SJ. (2002).
Cultivation of the ubiquitous SAR11 marine
bacterioplankton clade. Nature 418: 630–633.
Redfield AC, Ketchum BH, Richards FA. (1963). The
influence of organisms on the composition of seawater.
In: Hill MN (ed), Comparative and descriptive oceanography. Wiley: New York, NY, USA, pp 26–77.
HMW DOM selects for methylotrophs
OA Sosa et al
2739
Repeta DJ, Aluwihare LI. (2006). Radiocarbon analysis of
neutral sugars in high-molecular-weight dissolved
organic carbon: Implications for organic carbon
cycling. Limnol Oceanogr 51: 1045–1053.
Rich VI, Pham VD, Eppley J, Shi Y, DeLong EF. (2011).
Time-series analyses of Monterey Bay coastal microbial picoplankton using a ‘genome proxy’microarray.
Environ Microbiol 13: 116–134.
Saitou N, Nei M. (1987). The neighbor-joining method: a
new method for reconstructing phylogenetic trees. Mol
Biol Evol 4: 406–425.
Sarmento H, Gasol JM. (2012). Use of phytoplanktonderived dissolved organic carbon by different types of
bacterioplankton. Environ Microbiol 14: 2348–2360.
Sarmento H, Romera-Castillo C, Lindh M, Pinhassi J, Sala
MM, Gasol JM et al. (2013). Phytoplankton speciesspecific release of dissolved free amino acids and their
selective consumption by bacteria. Limnol Oceanogr
58: 1123–1135.
Schwalback MS, Tripp HJ, Steindler L, Smith DP,
Giovannoni SJ. (2010). The presence of glycolysis
operon in SAR11 genomes is positively correlated with
ocean productivity. Environ Microbiol 12: 490–500.
Sharma AK, Becker JW, Ottesen EA, Bryant JA, Duhamel S,
Karl DM et al. (2013). Distinct dissolved organic matter
sources induce rapid transcriptional responses in
coexisting populations of Prochlorococcus, Pelagibacter
and the OM60 clade. Environ Microbiol 16: 2815–2830.
Shieh WY, Liu TY, Lin SY, Jean WD, Chen JS. (2008).
Simiduia agarivorans gen. nov., sp. nov., a marine,
agarolytic bacterium isolated from shallow coastal
water from Keelung, Taiwan. Int J Syst Evol Microbiol
58: 895–900.
Shrestha PM, Nevin KP, Shrestha M, Lovley DR. (2013).
When is a microbial culture “pure”? Persistent cryptic
contaminant escapes detection even with deep genome
sequencing. mBio 4: e00591–12.
Song J, Oh HM, Cho JC. (2009). Improved culturability of
SAR11 strains in dilution-to-extinction culturing from
the East Sea, West Pacific Ocean. FEMS Microbial Lett
295: 141–147.
Sowell SM, Abraham PE, Shah M, Verberkmoes NC,
Smith DP, Barofsky DF et al. (2010). Environmental
proteomics of microbial plankton in a highly productive coastal upwelling system. ISME J 5: 856–865.
Stamatakis A. (2014). RAxML Version 8: A tool for
Phylogenetic Analysis and Post-Analysis of Large
Phylogenies. Bioinformatics 30: 1312–1313.
Stingl U, Cho JC, Foo W, Vergin KL, Lanoil B, Giovannoni SJ.
(2008). Dilution-to-extinction culturing of psychrotolerant planktonic bacteria from permanently ice-covered
lakes in the McMurdo Dry Valleys, Antarctica. Microb
Ecol 55: 395–405.
Stingl U, Desiderio RA, Cho JC, Vergin KL, Giovannoni SJ.
(2007a). The SAR92 clade: an abundant coastal
clade of culturable marine bacteria possessing
proteorhodopsin. Appl Environ Microbiol 73:
2290–2296.
Stingl U, Tripp HJ, Giovannoni SJ. (2007b). Improvements
of high-throughput culturing yielded novel SAR11
strains and other abundant marine bacteria from the
Oregon coast and the Bermuda Atlantic Time Series
study site. ISME J 1: 361–371.
Sun J, Steindler L, Thrash JC, Halsey KH, Smith DP, Carter AE
et al. (2011). One carbon metabolism in SAR11 pelagic
marine bacteria. Plos One 6: e23973.
Suzuki MT, Preston CM, Béjà O, De La Torre JR, Steward
GF, DeLong EF. (2004). Phylogenetic screening of
ribosomal RNA gene-containing clones in bacterial
artificial chromosome (BAC) libraries from different
depths in Monterey Bay. Microb Ecol 48: 473–488.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M,
Kumar S. (2011). MEGA5: molecular evolutionary
genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods.
Mol Biol Evol 28: 2731–2739.
Tamura K, Nei M, Kumar S. (2004). Prospects for inferring
very large phylogenies by using the neighborjoining method. Proc Natl Acad Sci USA 101:
11030–11035.
Weiner RM, Taylor LE II, Henrissat B, Hauser L, Land M,
Coutinho PM et al. (2008). Complete genome sequence
of the complex carbohydrate-degrading marine bacterium, Saccharophagus degradans strain 2-40T. PLoS
Genet 4: e1000087.
Williams PM, Druffel ERM. (1987). Radiocarbon in dissolved
organic matter in the central North Pacific Ocean. Nature
330: 246–248.
This work is licensed under a Creative
Commons
Attribution-NonCommercialNoDerivs 4.0 International License. The images or
other third party material in this article are included
in the article’s Creative Commons license, unless
indicated otherwise in the credit line; if the material
is not included under the Creative Commons license,
users will need to obtain permission from the license
holder to reproduce the material. To view a copy
of this license, visit http://creativecommons.org/
licenses/by-nc-nd/4.0/
Supplementary Information accompanies this paper on The ISME Journal website (http://www.nature.com/ismej)
The ISME Journal