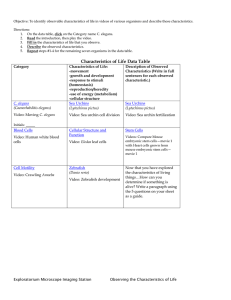
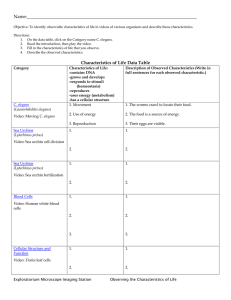
Genetic Analysis of the Timing, Morphology ... C. by
advertisement

Genetic Analysis of the Timing, Morphology and Degradation of Cell Corpses in C. elegans by Gillian Marie Stanfield A.B. University of Chicago, 1991 Submitted to the Department of Biology in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY June 1999 @ 1999 Gillian M. Stanfield. All rights reserved. The author hereby grants MIT permission to reproduce and to distribute paper and electronic copies of this thesis document in whole or in part. Signature of Author: Department of Biology May 25, 1999 A Certified by: CA xl 7~~> H. Robert Horvitz Professor of Biology Thesis Supervisor I-) Accepted by: Alan Grossman Chairman of the Graduate Committee Department of Biology 1 AR9 V LIBRARIES Genetic Analysis of the Timing, Morphology and Degradation of Cell Corpses in C. elegans Gillian M. Stanfield Abstract Programmed cell death is an important process during the development and life of multicellular organisms. The process by which many cell types undergo programmed cell death is morphologically and mechanistically similar and has been termed apoptosis. To understand how cells achieve this conserved cell-death fate, we have analyzed the genetic control of downstream events in the cell-death process in the nematode Caenorhabditiselegans. We have found that mutations in the gene ced-8 alter the kinetics of appearance of cell corpses during C. elegans embryogenesis and that ced-8 encodes a protein similar to human XK, a putative membrane transport protein that is implicated in a form of hereditary neuroacanthocytosis. We isolated loss-of-function mutations in a novel gene, ced-11, which alter the conserved morphology of programmed cell deaths. We found that CED-11 shares similarity with TRP calcium channels, suggesting that calcium signaling may be important for the downstream morphological events in apoptotic cells. To develop markers for studying downstream cell-death processes during programmed cell death, we developed methods to assay DNA degradation in C. elegans cell corpses. We examined the kinetics of DNA degradation in the wild type and in the DNA degradation-defective mutant nuc-1 and found that DNA degradation during programmed cell death involves multiple genetically-separable steps, not all of which are under the control of nuc-1. We found that NUC-1 is similar in sequence to mammalian DNase II, thus confirming a role for this endonuclease in the pathway for programmed cell death. Thesis Supervisor: H. Robert Horvitz Title: Professor of Biology 2 Acknowledgments I thank my advisor, Bob Horvitz, for his instruction, patience and encouragement, and the gift of freedom for exploration. I thank Sylvia Sanders, Frank Solomon and my committee, Richard Hynes, Christine Li, Terry Orr-Weaver and Hermann Steller, for sharing their insight and advice. I thank my teachers, especially Eric Jones (Biology I), Jacquelyn Beals (Genetics I), Jerry Coyne and Herbert Friedmann. I thank Dave Demirjian for introducing me to molecular biology at an impressionable time in my life, and Malcolm Casadaban and Lucia Rothman-Denes for letting me loose in their labs. I thank my collaborators Yi-Chun Wu, Erika Hartwieg, Nancy Tsung and Beth James for their help. I thank all the inhabitants of the Horvitz lab for being true lab comrades, for their advice and questions, and for conversations about science, movies, sports and other less-important things. I especially thank Shai Shaham for all those stimulating discussions in the Bay of Optimism, Tory Herman for sharing the Times crossword puzzle, and Xiaowei Lu for tolerating two teaching assignments with me. In addition, I thank Michael Basson, Andrew Chisholm, Yishi Jin, Beth Sawin and Erik Jorgensen for making my first years in the lab both interesting and enjoyable and Ewa Davison, Brad Hersh, Ho-Yon Hwang and Liz Speliotes for the same thing, only later. I thank my friends and classmates, especially Farhang, Jennifer, Sandy, Adam, Mark, Leila, Matt, and Monicia. Two people made this work possible. My mother Rhonda has always encouraged me to achieve what I wish and to love what I do, and I hope that she is proud of me. My husband Mark has shared everything along the way: nothing less can be said. 3 Table of Contents 1 Title p ag e .................................................................................................................................. Ab stract.....................................................................................................................................2 Acknowledgm ents...................................................................................................................3 4 Table of Contents ..................................................................................................................... 8 Chapter 1. Introduction: Apoptosis: Cell Killing and Downstream Events .......... 9 Overview .............................................................................................................................. 10 Introduction to program med cell death...................................................................... 10 The conserved features of apoptotic cell death ...................................................... Apoptotic programmed cell deaths occur in Caenorhabditiselegans.......................12 13 Control of program m ed cell death ............................................................................... Program med cell death requires activities within dying cells ................................ 13 The execution of programmed cell deaths................................................................14 14 Cell killing in C. elegans ............................................................................................ Cell killing in m am m alian cells..................................................................................16 16 Caspase regulation and cascades in mam malian cells............................................ 17 Mitochondria and cell killing.................................................................................... Many mammalian Bcl-2 family proteins, even more proposed functions............18 Downstream events of apoptosis......................................................................................19 Morphological and biochemical changes in the dying cell are triggered by 19 upstream events ............................................................................................................. 20 M any caspase substrates ............................................................................................ 20 M orphological and biochem ical changes in the dying cell ..................................... levels.......................................................................20 calcium intracellular Increased 21 Cytoskeletal rearrangements and loss of cell-cell contacts................................. 21 Phosphatidylserine exposure at the plasm a membrane ..................................... 22 Intracellular acidification of dying cells ............................................................... DNA degradation.......................................................................................................22 23 Cell-death endonucleases ..................................................................................... 26 Chrom atin condensation ....................................................................................... 27 ............................ activity transglutaminase tissue by Crosslinking of proteins Uncoupling of downstream cell-death events.............................................................27 28 Defects in downstream events can affect cell killing .................................................. Cell-corpse engulfm ent...............................................................................................28 28 Cell corpses are cleared from living tissues by engulfment ................ 29 Cell-corpse engulfment: possible recognition m echanism s ................................ 31 Cell-corpse engulfment: phagocytosis ................................................................. Engulfment and cell killing........................................................................................31 33 Conclusions ........................................................................................................................ Acknowledgm ents.............................................................................................................34 References...........................................................................................................................35 Chapter 2. The ced-8 gene controls the timing of programmed cell deaths 48 in C. elegans.................................................................................................... ary.............................................................................................................................49 Summ Introduction........................................................................................................................50 52 Resu lts ................................................................................................................................. The appearance of cell corpses is delayed in ced-8 mutant embryos.....................52 53 ced-8 has a weak effect on cell killing ........................................................................ 4 ced-8 acts dow nstream of or in parallel to ced-9...........................................................54 55 DN A degradation in ced-8 mutants .............................................................................. 57 Cloning of ced-8 .............................................................................................................. 58 Sim ilarity of CED -8 to XK and other proteins ......................................................... D iscussion...........................................................................................................................58 58 ced-8 m utations delay the appearance of cell corpses .............................................. 59 Function of ced-8 in program med cell death........................................................... 61 of CED -8......................................................................... conservation Evolutionary 63 Experim ental procedures .............................................................................................. N em atodes......................................................................................................................63 63 G enetic m apping ........................................................................................................... 63 Cell D eath A ssays .......................................................................................................... 64 ced-8 dosage experim ents .......................................................................................... Rescue experim ents........................................................................................................64 64 Molecular biology .......................................................................................................... cD N A analysis................................................................................................................65 67 A cknow ledgm ents ............................................................................................................. References...........................................................................................................................68 73 Tables .................................................................................................................................. 73 Table 1. ced-8 alleles................................................................................................... 74 Table 2. ced-8 enhances w eak ced-3 and ced-4 alleles .............................................. 75 ................ to ced-9 parallel of or in Table 3. ced-8 functions downstream Figures.................................................................................................................................76 Figure 1. Delayed appearance of programmed cell deaths in ced-8 animals ........ 76 Figure 2. Delayed onset of DNA degradation in ced-8 embryos..............78 Figure 3. ced-8 cloning and cDN A sequence...............................................................80 82 Figure 4. ced-8 developm ental northern blot. .......................................................... 84 Figure 5. CED -8 protein sequence com parisons..................................................... 86 Figure 6. ced-8:gfp expression ................................................................................... Chapter 3. ced-1 1 is required for the apoptotic morphology of programmed cell 88 deaths in C. elegans ......................................................................................... Results.................................................................................................................................93 93 A screen for cell-death m utants .................................................................................... Cell corpses appear abnormally non-refractile in ced-1 1 mutant embryos ............ 94 96 Engulfm ent of ced-1 1 non-refractile cell corpses .................................................... 97 ............................ by EM Non-refractile ced-1 1 cell corpses are not electron-dense ced-11 cloning..................................................................................................................98 99 CED -11 is w eakly sim ilar to TRP proteins................................................................ D iscussion.........................................................................................................................101 101 ced-11 controls cell-corpse m orphology ..................................................................... ced-1 1 acts downstream in the genetic pathway for programmed cell death.........102 CED -11 m ight function as a calcium channel............................................................103 M aterials and Methods....................................................................................................105 N em atodes....................................................................................................................105 105 Cell death assays .......................................................................................................... 105 Genetic m apping .......................................................................................................... Rescue experim ents......................................................................................................106 Electron m icroscopy.....................................................................................................106 107 Molecular biology ........................................................................................................ 5 108 Acknow ledgm ents ........................................................................................................... 109 ................. References.................................................................................................... 113 Tables ................................................................................................................................ Table 1. Mutation of ced-1 1 does not result in survival of cells that normally die...................................................................................................................113 Table 2. Mutations that prevent cell deaths are epistatic to ced-1I ......................... 114 Figures...............................................................................................................................115 Fig. 1. A visual screen for mutants with altered patterns of cell corpses in 115 C. elegans embryos ........................................................................................... Fig. 2. Refractile cell corpses are reduced in number and abnormally nonrefractile structures are present in ced-11; ced-5(n2098) embryos ................ 117 Fig. 3. Ced-11 phenotype............................................................................................119 Fig. 4. The engulfment of non-refractile ced-1I cell corpses is delayed by mutations that delay the engulfment of normal cell corpses.......................121 Fig. 5. Kinetics of cell corpse appearance in ced-1I mutants....................................123 Fig. 6. N om arski and electron m icrographs of cell corpses.....................................125 Fig. 7. ced-11 cloning....................................................................................................127 Fig. 8. ced-1I transcript................................................................................................130 Fig. 9. CED-11 protein.................................................................................................132 Fig. 10. Pathw ay m odel...............................................................................................135 Chapter 4. NUC-1, a C. elegans DNase II homolog, functions in an intermediate step 137 in DNA degradation within cells dying by programmed cell death ..... Summ ary...........................................................................................................................138 Introduction......................................................................................................................139 Results...............................................................................................................................142 142 TUN EL labels a subset of dying cells in C. elegans .................................................... nuc-1 embryos have more TUNEL-reactive nuclei than do wild-type embryos.........................................................................................................................142 144 nuc-1 encodes a DN ase II hom ologue ........................................................................ 146 nuc-1 m ay function w ithin dying cells ....................................................................... Completion of DNA degradation during programmed cell death may require engulfing cells...............................................................................................................147 The engulfment genes ced-1 and ced-7 are involved in the initiation of DNA degradation...................................................................................................................147 Discussion.........................................................................................................................148 148 Endonuclease activity in dying cells .......................................................................... 149 Regulation of N U C-1 ................................................................................................... 150 A dditional activities required for DN A degradation ............................................... 151 ................................................................................................ Experim ental Procedures N em atodes....................................................................................................................151 152 Genetic m apping .......................................................................................................... Com plem entation tests................................................................................................152 Cell-corpse counts........................................................................................................152 153 U N EL .......................................................................................................................... SYTO 11 staining..........................................................................................................153 154 M olecular biology ........................................................................................................ 154 Deletion library screening ........................................................................................... 155 Acknow ledgm ents ........................................................................................................... References.........................................................................................................................156 6 160 T ab les ................................................................................................................................ Table 1. TUNEL specifically labels dying cells.........................................................160 Table 2. TUNEL labels persistent DNA in nuc-1 anim als........................................161 Table 3. Mutations in ced-2, -5, -6 and -10 do not alter TUNEL staining................162 Table 4. Mutations in ced-1 and ced-7 reduce TUNEL staining ............................... 163 F ig u res...............................................................................................................................164 Figure 1. Wild-type and nuc-1 embryos show different TUNEL-staining patterns........................................................................................................164 Figure 2. nuc-1 cloning................................................................................................166 Figure 3. n3335 TUNEL staining is like that of nuc-1(e1392). .................................. 169 Figure 4. Unengulfed cell corpses in engulfment-defective mutants are TUNEL-positive..........................................................................................171 Figure 5. Unengulfed cell corpses stain with SYTO 11............................................173 Figure 6. Model for DNA degradation during programmed cell death in 175 C. elegans. ..................................................................................................... Appendix 1. Genetics of programmed cell death in C. elegans:: Past, present and 177 fu ture ............................................................................................................. Abstract.............................................................................................................................178 Introduction......................................................................................................................179 egl-1, ced-4, ced-3 and ced-9 are global regulators of programmed cell death in 179 C. elegans ........................................................................................................................... 182 Pathway to death ............................................................................................................. ced-9 and ced-4 have both protecting and killing activities...........................................185 187 Developm ental regulation of programmed cell death ................................................. death.......................................................................................................190 In the throes of 191 Beyond the Valley of the Shadow of Death ................................................................... 192 Conclusions ...................................................................................................................... 194 Acknowledgements ......................................................................................................... F igures...............................................................................................................................198 Figure 1. Distribution of programmed cell deaths within the cell lineage of the C. elegans herm aphrodite..............................................................................198 Figure 2. Interactions am ong egl-1, ced-9, ced-4 and ced-3 ........................................ 200 Figure 3. A model for the activation of programmed cell death in C. elegans........203 7 Chapter 1 Introduction Apoptosis: Cell Killing and Downstream Events 8 Overview Programmed cell death is an important part of the development and homeostasis of all multicellular organisms. In mammals, programmed cell death is synonymous with apoptosis, which denotes a particular morphology of the dying cell. The mechanisms by which cells die are similar in animals as diverse as nematodes and mammals, consistent with the conservation of ultrastructural changes in dying cells. The downstream events of programmed cell death comprise the conserved morphology of dying cells and the engulfment and degradation of cell corpses. It appears that to some extent, the downstream events are activated independently of one another by the upstream regulators of cell death, caspases and mitochondria. 9 Introduction to programmed cell death It has become apparent that the genetically-controlled process of cell birth by cell division in multicellular animals is balanced by a genetic program of cell elimination, programmed cell death. During the development and life of many, if not all, multicellular animals, some cells are generated that perform only a transient function before being eliminated from living tissues, and other cells are generated that perform no function at all (reviewed by Vaux and Korsmeyer, 1999). In some tissues, cells are generated in excess and proper morphogenesis requires the elimination of part of the cell population (reviewed by Jacobson et al., 1997). For instance, programmed cell death can be used to accomplish stage-specific differences in morphology, as in the elimination of the amphibian tail bud, a structure that is useful to the tadpole but not to the adult frog. In other cases, programmed cell death is necessary for the elimination of structures that are formed but ultimately serve no purpose: during vertebrate development, interdigital webbing is formed, but is eliminated in animals with toes and fingers. As another example, both male and female genital primordia are initially formed but one regresses in response to hormonal cues upon the sex determination decision. Finally, the elimination of cells may be important for the maintenance of a well-functioning adult tissue. Many neural cells die during the development of the mammalian nervous system as a by-product of the process of target matching (reviewed by Pettmann and Henderson, 1998). During mammalian immune system development, any potentially self-reactive or non-functional pre-immune cells are eliminated, and failure of this process leads to disease (reviewed by Nagata, 1996). Some adult tissues are responsive to environmental or hormonal cues and can expand or contract according to need. If the proper balance between cell birth and cell death is not maintained, disease can result, either from too many cells (e.g., cancer) or too few (e.g., neurodegenerative disease) (reviewed by Thompson, 1995). The conserved features of apoptotic cell death In many of these instances of developmentally programmed cell death, the dying cells appear very similar. The term that is used to distinguish developmentally programmed cell death with this characteristic morphology is apoptosis (Kerr et al., 1972; Wyllie et al., 1980), defined in part as being distinct from necrosis, death that occurs as a result of cellular damage resulting in a loss of homeostasis. Morphological 10 changes in cells undergoing apoptosis comprise alterations in most aspects of the structure of the dying cell and many of these alterations can be visualized by light or electron microscopy. The dying cell shrinks, blebbing of the plasma membrane occurs, the cytoplasm and nucleus condense, the nuclear envelope becomes dilated, chromatin appears condensed, and the dying cell breaks up into smaller membrane-bound fragments. By contrast, organelles remain grossly intact at the initial stages of cell death. As visualized by EM, the dying cell becomes more darkly-staining ("electrondense") relative to most living cell types. Other biochemical changes of the dying cell can be detected. Phospholipids are normally asymmetrically distributed in the plasma membrane, and this asymmetry is lost in dying cells. For instance, phosphatidylserine (PS) becomes exposed on the outer leaflet of the membrane, as visualized by labeling with Annexin V, a calcium-dependent PS-binding protein (Vermes et al., 1995). Some vital dyes (e.g., acridine orange) are excluded from living cells but become concentrated within cell corpses (Saunders et al., 1962; Abrams et al., 1993). Condensed chromatin can be observed in fixed preparations or in living animals treated with DNA-labeling dyes. Increases in calcium concentration can be visualized within dying cells (Kaiser and Edelman, 1977). Proteins are crosslinked by tissue transglutaminase activity (reviewed by Melino and Piacentini, 1998). Although as visualized by EM mitochondria appear grossly intact, they undergo loss of transmembrane potential associated with opening of the permeability transition pore (PTP) and release of pro-apoptotic factors into the cytoplasm (reviewed by Susin et al., 1998). A long-recognized hallmark of apoptosis is the degradation of DNA by endonucleolytic activity (Wyllie, 1980). Analysis of the DNA of dying cells on agarose gels has shown that DNA is cleaved first to 50-100 kb fragments (Oberhammer et al., 1993b) and then at internucleosomal linker regions resulting in a fragment ladder (Wyllie, 1980; Oberhammer et al., 1993b). This partially-degraded DNA can also be detected using the TUNEL (terminal transferase-mediated dUTP nick end labeling; also called ISEL, in situ end labeling, and other names) technique, which labels free 3'hydroxyl DNA ends in fixed tissue (Gavrieli et al., 1992). Engulfment is triggered rapidly by cells dying by apoptosis. The cell corpse is engulfed and cleared from the living tissue by phagocytic cells, and the final 11 breakdown and recycling of the components of the dying cell occur within the phagocyte. Apoptotic programmed cell deaths occur in Caenorhabditiselegans Programmed cell death is a common cell fate in the nematode Caenorhabditiselegans: in the hermaphrodite, of 1090 somatic cells generated, 131 die (Sulston and Horvitz, 1977; Kimble and Hirsh, 1979; Sulston et al., 1983), and approximately half of the cells generated in the germline die (Gumienny et al., 1999). The lineal identities of dying cells in the soma are essentially invariant from animal to animal. Some cells may die, as in other organisms, to promote developmental events, e.g., morphogenesis, sex-specific differentiation and nervous system development (Ellis et al., 1991b). Few cells seem to perform any function before dying. However, the extra cells generated in the germline have been proposed to function analogously to nurse cells in the germline of other organisms (Gumienny et al., 1999). C. elegans programmed cell deaths are morphologically similar to those in other animals (Robertson and Thomson, 1982). Most of the ultrastructural changes that are observed by EM are conserved in C. elegans cell corpses, although the fragmentation of the cell corpse into smaller membrane-bound bodies does not occur. The DNA degradation process has not been analyzed using gel electrophoresis, but DNA of dying cells does become condensed and eventually disappears as assayed by Feulgen or DAPI staining (Hedgecock et al., 1983). TUNEL can be used to label C. elegans cell corpses (Wu et al., manuscript in preparation; see Chapter 4). Transglutaminase activity is present in C. elegans (Madi et al., 1998). An increased level of crosslinked protein is present in engulfment mutants, where cell corpses persist and therefore more are present, suggesting that transglutaminase indeed is active in dying cells. Assays for pH changes, calcium concentration within dying cells and surface phosphatidylserine exposure have not yet been used to examine C. elegans cell corpses. As in other organisms, C. elegans programmed cell deaths are cleared away by engulfment and degraded within the engulfing cell. However, there are no professional phagocytic cells in C. elegans; instead, neighboring cells are responsible for phagocytosis. It is thought that most cell types apart from neurons are capable of engulfment. 12 C. elegans biology offers several advantages for the study of the genetic control of programmed cell deaths. Since the identity of dying cells is the same in each individual, detailed analysis of the genetic control of the cell-death fate choice and the subsequent morphological and biochemical changes occurring in the dying cell can be performed. In theory, mutations affecting the cell death fate in any subset of cells within the animal can be identified and analyzed. Indeed, mutations affecting diverse aspects of the programmed cell death fate in all or a subset of cells have been identified and characterized (reviewed by Metzstein et al., 1998). Control of programmed cell death Programmed cell death requires activities within dying cells Several lines of evidence indicate that cell death requires gene function within dying cells. First, the nature of the morphological changes, such as the condensation of various cellular components and DNA degradation, that occur prior to engulfment by neighboring cells, suggest that proteins within the dying cell are actively promoting at least some aspects of the death process. Many genes that mediate the execution of programmed cell death have been identified, and some of these genes have been shown to function cell-autonomously. For instance, in C. elegans, the activities of the ced-3, ced-4, and egl-1 genes have been shown to be required for programmed cell death and these functions are required cell-autonomously; cell-specific loss of ced-3 or ced-4 function in mosaic animals prevents cell death, and cell-specific expression of ced-3, ced-4 or egl-1 is sufficient to induce the death of the expressing cells (Ellis and Horvitz, 1986; Yuan and Horvitz, 1990; Shaham and Horvitz, 1996; Conradt and Horvitz, 1998). In Drosophila,loss of rpr, hid, and grim functions eliminates most developmental programmed cell deaths; each of these genes can also induce cell-autonomous programmed cell death when overexpressed (White et al., 1994; Grether et al., 1995; Chen et al., 1996). Many genes that cal induce the death of mammalian cells have been identified, and some of these genes (e.g., caspase-3, caspase-9, Apaf-1) have been shown to be of functional importance in knockout mouse studies (Kuida et al., 1996; Cecconi et al., 1998; Kuida et al., 1998; Yoshida et al., 1998). Previous arguments for and against the role of active genes within dying cells focused primarily on studies showing that various cell types either would, or would not, undergo programmed cell death in the presence of protein synthesis inhibitors such 13 as cycloheximide (e.g., Wyllie et al., 1984; Martin et al., 1990). The conflict between these results is likely, in hindsight, to have resulted from differences in the levels of proand anti-apoptotic proteins present in these cells prior to treatment; many candidates now exist for molecules that may influence the sensitivity of cells to cell-death triggers. The execution of programmed cell deaths A description of cell-death execution in C. elegans offers a much-simplified overview of the classes of genes involved in programmed cell death, yet includes many of the best-understood classes of cell death execution genes; thus it provides a framework for understanding what is known about cell death generally. Therefore, we describe first the cell death pathway in the worm and second studies in other systems, primarily mammalian cells, that confirm the worm cell-death pathway and also indicate the existence of other mechanisms for cell killing. Details of the genetic analysis that has led to the pathway for programmed cell death execution in C. elegans are described in more detail in Appendix 1 (published as Metzstein et al., 1998). Cell killing in C. elegans The activities of three genes, egl-1, ced-4 and ced-3, are required for the execution of developmental programmed cell deaths in C. elegans (Ellis and Horvitz, 1986; Conradt and Horvitz, 1998). The activity of a fourth cell-death execution gene, ced-9, is required to prevent the deaths of cells that normally live (Hengartner et al., 1992). The activities of ced-3 and ced-4 are required for cell deaths caused by loss of function (lf) of ced-9, indicating that ced-9 normally functions to negatively regulate ced-4 and ced-3 activity in cells that should not die (Hengartner et al., 1992). By contrast, a loss-of-function allele of egl-1 does not suppress ced-9(lf) deaths, suggesting that egl-1 acts upstream of ced-9 (Conradt and Horvitz, 1998). Overexpression studies have been used to order the functions of ced-4 and ced-3 and indicate that ced-4 functions upstream of ced-3 (Shaham and Horvitz, 1996). These data suggest that when cells die, egl-1 inhibits ced-9 activity, allowing ced-4 to activate cell killing by ced-3, which appears to play the critical downstream role in effecting cell death. Molecular studies have confirmed these roles for these genes and have suggested that in fact the proteins may directly interact. egl-1 and ced-9 each encode Bcl-2 family proteins (Hengartner and Horvitz, 1994; Conradt and Horvitz, 1998); EGL-1 is a BH314 only (Bcl-2 homology 3) member of this family. Four BH domains (BH1, BH2, BH3 and BH4) have been defined, and all Bcl-2-family proteins have at least one of these domains; BH3-only proteins have pro-apoptotic activities (Adams and Cory, 1998). CED-4 is similar to mammalian Apaf-1 (apoptotic protease activating factor) (Zou et al., 1997), which was identified biochemically as a factor required for cell death in an in vitro system. ced-3 encodes a founding member of the caspase (cysteine aspartatespecific protease) family of proteases (Yuan et al., 1993). Caspases are synthesized as proenzymes and are processed via cleavage, frequently by other caspases, and assemble as homodimers of a heterodimer consisting of the approximately 13 kD and 15 kD subunits generated by cleavage (reviewed by Thornberry and Lazebnik, 1998). Some caspases, including CED-3, have long "prodomain" N-termini, which do not form part of the active enzyme but may be important for activation or other regulation (Thornberry and Lazebnik, 1998 and S. Shaham, B. Davies, P. Reddien and H.R.H., unpublished results). Based on binding and localization studies on the C. elegans proteins and their mammalian counterparts, a molecular model for cell-death execution has been developed. In living cells, CED-9/Bcl-2 is localized at the mitochondrial outer membrane, where it binds to and inhibits the activity of CED-4/Apaf-1. Death is initiated by activation, possibly by transcriptional upregulation, of EGL-1, which binds to CED-9, releasing CED-4. CED-4 then can bind to CED-3, thereby inducing processing and formation of the active enzyme. Additional caspase family members, csp-1, csp-2 and csp-3, recently have been identified in C. elegans genomic sequences (Shaham, 1998). These three genes encode at least six different proteins by alternative splicing, some of which have been shown to be capable of functioning biochemically as caspases. The csp genes have not yet been analyzed genetically and their role, if any, in programmed cell death has not been established. However, strong loss-of-function mutations in ced-3 lead to the survival of most but, significantly, not all cells that normally die in C. elegans, so the csp genes may be responsible for the residual killing activity observed in these animals. Another possibility is that the csp genes might function downstream of and be activated by ced-3 to activate a subset of killing functions in dying cells. Alternatively, the csp genes might function in processes other than programmed cell death. 15 Cell killing in mammalian cells As noted above, homologs of the four major cell death-execution genes in C. elegans have been identified in mammals. Mammalian data are generally consistent with the pathway described in C. elegans, and suggest that caspase activation and inhibition are mediated by protein-protein interactions. However, egl-1, ced-9 and ced-3 each are exemplified by multiple family members, leading to a much more complex picture of cell-death regulation. Furthermore, a mitochondria-dependent cell-death pathway, which interfaces with the caspase pathway at least in part, has been identified (reviewed by Kroemer et al., 1998). Caspase regulation and cascades in mammalian cells So far, at least fourteen different mammalian caspases have been identified (Thornberry and Lazebnik, 1998; Van de Craen et al., 1998). These are classified into three groups based on the criteria of structure, (probable) function and substrate specificity. One group (typified by caspase-1 /ICE) is probably involved in immune function rather than programmed cell death. The other proteins are divided into the categories of "initiator" and "effector" caspases. It is thought that procaspases are already present in many living cell types and that upon induction of cell death, initiator caspases (e.g., caspase-8/FLICE) are activated by an upstream signal (for instance, CED-4-like molecules as discussed for C. elegans). Active initiator caspases go on to cleave and thus activate effector caspases (e.g., caspase-3/CPP32), which are necessary for cell killing. However, substrate specificity and kinetic data of cleavage during the death process indicate that a unique pathway of linear caspase activation does not exist, and there can be feedback from effector to initiator caspases (reviewed by Thornberry and Lazebnik, 1998). In one of the best-understood models of cell-death initiation, cell death induced by TNF (tumor necrosis factor) binding leads to clustering of TNF receptor molecules, thus causing the association of procaspase molecules via "adaptor" proteins that bridge the cytoplasmic domain of TNFR and caspase-8 (reviewed by Kidd, 1998). Oligomerizing of caspases has been shown to be sufficient to drive processing without association with other proteins (Martin et al., 1998), so the general theme of using protein-protein interactions to drive association between and autoprocessing of caspase molecules might be used to initiate cell death induced by many signals. 16 Caspases are thought to be widely important for programmed cell death, since blocking their function prevents programmed cell death induced by a wide variety of stimuli (Thornberry and Lazebnik, 1998). Overexpression of p35, a baculovirusencoded caspase inhibitor, or other viral inhibitors (reviewed by Kidd, 1998) inhibits programmed cell death in many contexts (e.g., Hay et al., 1994; Sugimoto et al., 1994; Bump et al., 1995). Peptide inhibitors (e.g., Z-VAD.fmk), which bind to the caspase substrate-recognition site, also block programmed cell death. Inhibitors specific to certain caspases are also used to test the dependence of cell deaths on caspase function and to order the action of particular caspases, though not always with consistent results (e.g., Hirata et al., 1998; Slee et al., 1999), and the precise order of activation of particular caspases may be cell type- or death stimulus-specific. Mitochondria and cell killing A class of models of cell-death activation focuses on various aspects of the loss of mitochondrial homeostasis and the concomitant release of cell-killing activating molecules, cytochrome c and AIF (apoptosis-inducing factor), from the mitochondria. Cytochrome c first was implicated in programmed cell death as one of three components purified from HeLa cell extracts that were necessary and sufficient, along with ATP, for cell death in an in vitro model (Liu et al., 1996). All three of these proteins, Apaf-1, -2 and -3 (agoptosis-activating factor), have now been characterized molecularly, and their identity provides a possible link between the caspase- and mitochondria-oriented models of cell death. Apaf-1 is similar in part to CED-4, but contains an additional WD repeat domain (Zou et al., 1997). Apaf-2 is cytochrome c (Liu et al., 1996). Apaf-3 is a caspase, caspase-9 (Li et al., 1997). Apaf-1, ATP and the cytochrome c are thought to function in activation of caspase-9 (Li et al., 1997; Zou et al., 1997). Thus, it is possible that in at least some cell types, the primary effect of the loss of mitochondrial homeostasis is to release cytochrome c into the cytoplasm, allowing it to potentiate the CED-4/Apaf-1-mediated activation of caspases. Furthermore, some cell death induced by cytochrome c is blocked by caspase inhibitors. However, cytochrome c is not necessarily released into the cytoplasm during apoptosis in all cells; for instance, it is not released in the death of multiple myeloma cells induced by Fas or dexamethasone (Chauhan et al., 1997). 17 AIF, which is similar in sequence to bacterial oxidoreductases, was identified as a mitochondrial factor capable of inducing nuclear apoptosis (Susin et al., 1999). How it functions to promote cell death is not known. When first identified as a factor released from the mitochondria during cell death, AIF appeared to be blocked by caspase inhibitors (Susin et al., 1996). More recently, the sequence of AIF has been reported and now AIF-induced death is not blocked by the peptide inhibitor Z-VAD.fmk, so it may be caspase-independent (Susin et al., 1999). A reduction of mitochondrial transmembrane potential associated with opening of the permeability transition pore (PTP) has been observed in dying cells. How are mitochondria regulated to release proapoptotic proteins? While cytochrome c and AIF might exit the mitochondria via the PTP, it has been shown that cytochrome c release is independent of the permeability transition (Kluck et al., 1997; Yang et al., 1997) and caspases can activate the permeability transition (Susin et al., 1997). Thus, caspases and the mitochondrial killer proteins can promote one another's activity. It is possible that neither mitochondrial permeability transition nor caspase activation should be regarded as the only key event in cell killing and that these two events normally both occur and are important in dying cells. There is as yet no evidence either in favor of or against a role of mitochondria during programmed cell death in C. elegans. A factor such as cytochrome c, which is required for viability, would not have been identified in most genetic screens for celldeath mutants (Metzstein et al., 1998). As a first test of whether cytochrome c or the mitochondrial permeability transition might play a role in programmed cell death in C. elegans, one could examine mitochondrial function or cytochrome c localization in cells programmed to die (Varkey et al., 1999). Many mammalian Bcl-2 family proteins, even more proposed functions Many Bcl-2 family proteins have been identified in mammals (reviewed by Adams and Cory, 1998; Green and Reed, 1998). Both pro-apoptotic and anti-apoptotic family members have been identified, and these proteins can self-associate as homodimers or bind one another in various heterodimeric combinations. Many Bcl-2-family proteins are found associated with cell membranes, particularly of the mitochondria, and also of the endoplasmic reticulum and nuclear envelope. The majority of models of Bcl-2 family function focus on their localization at the mitochondria (Reed et al., 1998). First, 18 as described above for C. elegans CED-9, anti-apoptotic Bcl-2 family proteins localized to the mitochondria might bind and inhibit Apaf-1. Second, some family members have been found to be capable of forming channels (Minn et al., 1997; Schendel et al., 1997; Schlesinger et al., 1997) that may be involved in ion flux between the mitochondria and cytoplasm and thereby regulate mitochondrial homeostasis. Third, Bcl-2 family members may modulate either release of cytochrome c and other pro-apoptotic factors from the mitochondria or opening of the PTP (Cai et al., 1998). Overexpression of Bcl-2, which blocks cell death, also blocks the release of cytochrome c (Yang et al., 1997). Many other functions have been suggested for Bcl-2 family proteins. It appears that both the pro- and anti-apoptotic Bcl-2 proteins may be active; that is, neither exists solely to antagonize the function of the other (e.g., Knudson and Korsmeyer, 1997). Downstream events of apoptosis Morphological and biochemical changes in the dying cell are triggered by upstream events Caspases are thought to be the major regulators of the "downstream events" of cell killing. We define these downstream events as the morphological changes in the dying cell, the engulfment of the cell corpse, and the degradation of the DNA and other components of the cell corpse. Each of these aspects of cell death by itself is probably not required for the execution of cell deaths, but all are conserved aspects of the celldeath process. Furthermore, it seems likely that blocking multiple downstream functions would inhibit cell-death execution, although blocking any one downstream function might not be. Since inhibition of caspase activity blocks cell death, including all signs of morphological changes, in most cell types (Thornberry and Lazebnik, 1998), caspases probably activate the various downstream events of programmed cell death. Cleavage by caspases probably activates some proteins and inactivates others in order to accomplish the downstream events of cell death. Increasing attention is also being paid to modes of cell death that are not blocked by caspase inhibitors and thus may be independent of caspase activity (Kawahara et al., 1998; Samejima et al., 1998; Susin et al., 1999). While the caspases have defined proteolytic activities that are required for their cell-killing functions, it remains to be determined which are the critical substrates that must be cleaved to effect cell death. It 19 is not clear how apoptotic downstream events might be effected in cells dying by caspase-independent mechanisms. Many caspase substrates One approach to identifying molecules that may be important for cell-death regulation or downstream cell-death events is to search for caspase substrates. Thus far, more than 60 proteins that are cleavable in vitro have been identified (reviewed by Stroh and Schulze-Osthoff, 1998), and some of these proteins have been shown to be cleaved during cell death. However, the function of many of these proteins in cell killing is unclear. A large fraction of these candidate caspase targets function in cell cycle regulation or signal transduction. Most of these targets were not identified in unbiased screens, but it is reasonable that a number of targets inactivated by the cell-death machinery should be molecules that promote cell division or non-death-related signal transduction. Unfortunately, it is difficult to test the hypothesis that most of the identified caspase substrates, many of which are important in living cells for viability of the animal, are important for apoptotic cell-death execution. Morphological and biochemical changes in the dying cell Increased intracellular calcium levels Increases in cell calcium can be observed in dying cells (Kaiser and Edelman, 1977). Compounds that increase intracellular Ca2" (e.g., Ca 2 +ionophores, thapsigargin) can induce cell death (Kaiser and Edelman, 1978; Jiang et al., 1994). Because of the wide variety of activities modulated by Ca 2 , in the cell, it is difficult to determine which of its targets are important for apoptosis and, more generally, whether Ca 2 , induces or potentiates cell death or whether Ca 2 , flux is merely a byproduct of cell killing. For instance, while Ca 2, increases can induce the mitochondrial permeability transition, Ca 2 . is also released from mitochondria during the permeability transition ( reviewed by Zoratti and Szabo, 1995). Calcineurin activity may be important for Ca 21-induced cell death (Shibasaki and McKeon, 1995). Recently, activated calcineurin has been shown to dephosphorylate BAD, a pro-apoptotic Bcl-2-family protein, thereby inducing its binding to and presumably inhibition of anti-apoptotic bcl-xL (Wang et al., 1999). As described below, Ca 2 + also may promote downstream events by modulating the activity of downstream cell-death factors. 20 Cytoskeletal rearrangementsand loss of cell-cell contacts Alterations in the cytoskeleton occur during cell death and may participate in membrane blebbing, cellular shrinkage and the formation of apoptotic bodies. Many cytoskeletal structural and regulatory proteins are substrates for caspases (Stroh and Schulze-Osthoff, 1998). For instance, cleavage of 19-catenin occurs during cell death, and the cleavage product can no longer bind a-catenin, suggesting that this cleavage should interfere with the ability of the cell to organize actin at sites of cell contact (Brancolini et al., 1997). Similarly, cleavage of pp125FAK should promote disassembly of focal contacts (Levkau et al., 1998). In addition, actin, fodrin, Gas2 and keratins are caspase substrates and are probably inactivated by cleavage (Stroh and Schulze-Osthoff, 1998). By contrast, gelsolin cleavage by caspases results in a constitutively active form of the protein, which may function to alter the structure of the cytoskeleton (Geng et al., 1998). Expression of the cleavage fragment of gelsolin resulted in rearrangement of the cytoskeleton, rounding up of cells and nuclear fragmentation, and dying gelsolin -/neutrophils have a delayed onset of membrane blebbing and DNA fragmentation (Kothakota et al., 1997). Caspase cleavage of the p21-activated kinases, PAK2 and hPAK65, results in constitutively active kinases, which could be involved in altering the cytoskeletal structure of the dying cell. Expression of the cleavage product of hPAK65 in culture cells resulted in apoptosis-like morphological changes: cell and nuclear shrinkage and chromatin condensation (Lee et al., 1997). Expression of a dominant negative PAK2 in cells dying by Fas-mediated killing prevented the fragmentation of dying cells to form apoptotic bodies, but enhanced exposure of PS on the cell surface; other morphological changes of cell death occurred on time in these cells (Rudel and Bokoch, 1997). Phosphatidylserineexposure at the plasma membrane Phospholipids are normally asymmetrically distributed within plasma membranes: phosphatidylserine (PS) is normally confined to the inner membrane leaflet; phosphatidylethanolamine (PE) is primarily in the inner leaflet; and sphingomyelin and phosphatidylcholine (PC) are primarily in the outer leaflet. This asymmetry is maintained by the action of lipid translocases within the membranes that generate and maintain membrane asymmetry (reviewed by Zwaal and Schroit, 1997). A scramblase 21 activity, which can promote bidirectional non-specific mixing of lipids within the membrane, originally was postulated to account for the observations that calcium increases the rate of lipid mixing (Bevers et al., 1982) and that diffusion within the membrane does not account for the degree of lipid mixing observed in cells dying by apoptosis or cells in which translocase activity is blocked. A calcium-dependent scramblase now has been further biochemically characterized (Basse et al., 1996) and molecularly cloned (Zhou et al., 1997). In dying cells, PS becomes exposed on the outer membrane leaflet; this phenomenon has been shown by a number of criteria including Annexin V binding to the surface of dying cells (Fadok et al., 1992b; Martin et al., 1995; Vermes et al., 1995). Calcium influx from the extracellular medium is important for PS exposure in dying cells (Hampton et al., 1996). It is possible that PS exposure is linked to the rise in calcium levels that occurs during apoptosis via the effects of calcium, which should activate scramblase activity and inhibit translocase activity (reviewed by Fadok et al., 1998). Intracellular acidification of dying cells Cellular homeostasis is maintained normally by ion transporters that regulate the concentration of various ions within the cell. Dying cells have been reported to exhibit intracellular acidification (e.g., Barry and Eastman, 1992; Gottlieb et al., 1996; Meisenholder et al., 1996), which is blocked by Bcl-2 overexpression or caspase inhibitors (Meisenholder et al., 1996; Wolf and Eastman, 1999). An alteration in the setpoint of the Na'/H' antiporter has been suggested to account for the acidification of cells undergoing programmed cell death (Li and Eastman, 1995). In addition, acidification can be delayed by upregulation of the vacuolar H*/ATPase (Gottlieb et al., 1995b). Conversely, inhibition of the vacuolar H*/ATPase induces p53-dependent apoptosis (Long et al., 1998). One possible mechanism of regulation of the Na*/H' antiporter is binding by Ca 2*/calmodulin, which reduces regulation by a pH-sensing domain in the antiporter (reviewed in Wakabayashi et al., 1997). DNA degradation DNA degradation can be assayed by gel electrophoresis or TUNEL. While electrophoretic analysis requires sampling a population of dying cells, TUNEL assays allow the detection of degraded DNA in single dying cells. The correspondence 22 between laddering and TUNEL reactivity is not completely clear, but the timing of the appearance of TUNEL-reactive cells correlates with the appearance of DNA laddering (Gavrieli et al., 1992), suggesting that TUNEL may detect laddered DNA. However, the minimum number of DNA ends required for detection of a dying cell by the TUNEL method is not known, and it is not clear at what point degrading DNA is no longer TUNEL-reactive. TdT and other polymerases used for in situ labeling assays utilize 3'hydroxyl ends as substrates; since these methods label cell corpses, it seems that endonuclease activities involved in degradation of cell-corpse DNA should generate such ends. The DNA of HeLa cell nuclei incubated with extracts from apoptotic thymocytes was cleaved into large 30-50 kb fragments and laddered fragments, both of which had 3'-hydroxyl termini, as assayed by end-labeling after gel electrophoresis (Hughes et al., 1998). However, it is possible that other enzymes such as phosphatases may be active in the nuclei of dying cells and may modify the end structure of the DNA. DNA degradation, like other downstream cell-death events, is coupled with celldeath execution: blocking components of the execution machinery, e.g., by inhibition of caspases or overexpression of anti-apoptotic Bcl-2 family proteins, also prevents detectable DNA fragmentation. However, mutations in caspase-3 (CPP32) have revealed a specific role for this caspase in DNA degradation. Some cell types lacking caspase-3 can undergo programmed cell death, but these cells fail to undergo chromatin condensation and DNA degradation when they die; most other aspects of cell-death morphology are normal (Janicke et al., 1998; Woo et al., 1998; Zheng et al., 1998). Cell-death endonucleases Many candidate cell-death endonucleases have been identified. Many studies have implicated a Ca2+, Mg 2+-dependentendonuclease as being important for DNA fragmentation (reviewed by Hughes and Cidlowski, 1994). If a single endonuclease is involved, the consensus candidate should be Ca2+- and Mg 2+-dependent,be localized to the nucleus of dying cells, produce 3'-hydroxyl ends and internucleosomal fragmentation, and be activated in a caspase-dependent way by the execution process (Hughes and Cidlowski, 1994). DNase I is Ca2+- and Mg 2+-dependent and nicks DNA to produce 5'-phosphate and 3'-hydroxyl ends. Incubation of T-cell hybridoma nuclei with serum containing DNase I induced internucleosomal fragmentation (Peitsch et al., 1992; Peitsch et al., 1993). 23 Increased levels of DNase I have been observed in tissues with high numbers of programmed cell deaths, such as prostrate gland of castrated rats (Kyprianou et al., 1988). DNase II, or the acidic endonuclease, is an abundant nuclease that is active at low pH (lower than approximately 6.2 in vitro). It was implicated as a possible cell-death endonuclease by Barry and Eastman (1993), who incubated CHO cell nuclei at various pH conditions and in the presence or absence of Ca2+ or Mg 2+ and found that DNA digestion occurred at acidic pH. They purified a nuclease with identical properties to DNase II from these cells and found that it was present in the nuclear fraction of both apoptotic and non-apoptotic cells and that it induced DNA laddering. Dying neutrophils were also found to contain a DNase 11-like activity (Gottlieb et al., 1995a). DNase II has been localized to the nuclei of lens fibers, which undergo an apoptosis-like degeneration (Torriglia et al., 1995). Finally, transfection of CHO cells with DNase II induces DNA condensation and apoptosis (Krieser and Eastman, 1998). DNase II meets some but not all criteria to be the cell-death endonuclease. Intracellular acidification, which may occur in dying cells (Barry and Eastman, 1992), represents a means by which DNase II might be selectively activated. However, DNase II activity is neither Ca2+- nor Mg2+-dependent, and it produces 3'-phosphate and 5'-hydroxyl ends, so it should not generate TUNEL-reactive DNA. It is not known whether DNase II can be activated by caspases. Thus, if it is involved in DNA degradation it is probably not the unique celldeath endonuclease (see Chapter 4). An 18 kD protein with calcium-dependent endonuclease activity was identified from rat thymocytes and designated NUC18 (Gaido and Cidlowski, 1991). Peptide sequences from purified NUC18 were found to be nearly identical to sequences from cyclophilin (Montague et al., 1994), so cyclophilins A, B and C were assayed and found to have endonuclease activity similar to that of the proposed cell-death endonuclease. This activity was Ca2+- and Mg 2+-dependent and produced 3'-hydroxyl DNA termini. Cyclophilin was found to be in the cytoplasmic and nuclear fractions of both apoptotic and nonapoptotic cells (Montague et al., 1997). When cyclophilin C was incubated with isolated HeLa cell nuclei, DNA was degraded to large (50kb) fragments (Montague et al., 1997), suggesting that cyclophilins may be involved in the generation of large fragments of DNA rather than in internucleosomal fragmentation. 24 A number of other novel endonucleases have been implicated in cell-corpse DNA degradation. Evidence for the involvement of such novel endonucleases in apoptosis was reviewed by Hughes and Cidlowski (1994). L-DNase II (Torriglia et al., 1998) was purified from porcine spleen and shown to be modified and upregulated in some apoptotic cells. It represents a cleaved form of leukocyte elastase inhibitor (LEI), a serpin, and its activity is similar to that of DNase II. Rat DNase Y (Liu et al., 1998) is similar in sequence (42% identity) and activity to DNase I and is localized to the nucleus. A complex of three nuclear proteins (Zhang et al., 1995), corresponding to a single endonuclease activity, termed NP42-50, was characterized from Jurkat cells and appeared to have an activity distinct from DNase I or DNase II. Nikonova et al. (1993) identified, in rat thymocytes, three distinct endonuclease activities, one Mn2'_ dependent, one acidic endonuclease-like, and one Ca 2 1- or Mn 2*-dependent. NUC70 is a 70-kDa endonuclease identified in human hematopoietic cells that is Ca 2 ' and Mg 2._ dependent and active in the pH range 6-8 (Urbano et al., 1998). NUC70 expression levels were similar in apoptotic cells and non-apoptotic cells, but activity was reduced in cells treated with peptide caspase inhibitors. The sequence of NUC70 has not been reported. While some or all of these endonucleases could be involved in programmed cell death, it remains to be seen whether they are functionally important and also how they are regulated by cell-death execution. Human DFF (_DNA fragmentation factor) was identified as an activity composed of 40 kD and 45 kD subunits required for nuclear apoptosis, including DNA fragmentation, in an in vitro cell-death assay (Liu et al., 1997). The 40 kD subunit was cloned and named CPAN (caspase-activated nuclease) (Halenbeck et al., 1998). The murine versions of the 45 kDa and 4- kDa proteins, CAD and ICAD (cgaspase-activated DNase, Inhibitor of CAD) were independently characterized and found to be activated directly by caspase activity (Enari et al., 1998; Sakahira et al., 1998). In living cells, CAD and ICAD exist as a complex. When caspases become active, ICAD is cleaved, releasing CAD to effect DNA degradation (Sakahira et al., 1998). However, in the absence of ICAD, CAD is not produced, suggesting that ICAD is required for CAD protein synthesis (Enari et al., 1998). CPAN/CAD functions as an endonuclease, although HMG2 may be required as a cofactor (Toh et al., 1998). In ICAD -/- mice, apoptotic deaths can occur, but cell corpses fail to undergo either DNA fragmentation, as visualized by TUNEL labeling, or condensation (Zhang et al., 1998). Thus, ICAD is 25 clearly important for DNA degradation in murine cells in vivo, probably because of its role in CAD synthesis, although the CAD knockout phenotype has not been analyzed. The other candidate cell-death endonucleases have not been analyzed genetically, and it remains to be seen whether they are truly involved in the DNA degradation process, or whether the evidence in favor of their involvement is simply correlative. Of course, even if loss of function of a particular endonuclease does not prevent DNA degradation in cell corpses, it is possible that other enzymes compensate for the lost activity. As described above, there are arguments for and against each of these candidates being the cell-death endonuclease, most of which are resolved trivially by acknowledging that multiple nucleases may be involved in the DNA-degradation process, or that different cell types may utilize slightly different pathways. C. elegans would provide an excellent model for testing the function of various candidate endonucleases during cell death. Cell death is dispensable during development, and a nuc-1 mutant, in which the degradation of DNA from ingested bacteria is blocked or at least greatly reduced, is viable. Essentially the entire genome sequence of the worm has been determined, and some, though not all of the candidate endonucleases are represented. For instance, three DNase II-like genes are predicted (Baker et al., 1998; Krieser and Eastman, 1998; Yasuda et al., 1998 and see Chapter 4). At least eleven cyclophilin genes are present (Page et al., 1996). However, CAD and ICAD apparently are not represented in the C. elegans genomic sequence, which is 99% complete. Chromatin condensation As cells die, their chromatin condenses, and the DNA is degraded. In cell deaths induced by TGF-191, chromatin condensation preceded the onset of detectable DNA degradation (Oberhammer et al., 1993a). However, loss of function of DFF45 blocks all nuclear changes with programmed cell death, not just DNA fragmentation, suggesting that fragmentation is required for condensation (Zhang et al., 1998). Therefore, either DFF45 has a second function in DNA condensation, or DNA fragmentation is required for condensation but not necessarily at a level detectable by the TUNEL assay. Hendzel et al. (1998) examined the condensation state of apoptotic nuclei using antibodies against various histones, and found that this DNA condensation was distinct from 26 mitotic condensation and that euchromatin was rapidly degraded, whereas heterochromatin aggregated to form pycnotic nuclei. Crosslinkingof proteins by tissue transglutaminaseactivity Tissue transglutaminase (tTG) catalyzes the conjugation of polyamines to proteins and the formation of isopeptide bonds between glutamine and lysine residues within proteins, resulting in covalent crosslinks (reviewed by Melino and Piacentini, 1998). tTG is positively regulated by Ca". The activity of tTG increases in dying cells and this activity correlates with the formation of SDS-resistant apoptotic bodies (Fesus et al., 1989). The level of tTG-catalyzed crosslinks in various cell death-defective mutants in C. elegans has been examined: while an increased level of crosslinked protein was observed in engulfment-defective animals, a decreased level was seen in executiondefective animals, showing a correlation between corpses and crosslinking (Madi et al., 1998). Transfection of tTG into 3T3 cells resulted in membrane blebbing and shedding of apoptotic-like bodies (Gentile et al., 1992). tTG-catalyzed crosslinking may function to stabilize the cell corpse and thereby prevent lysis prior to engulfment. Uncoupling of downstream cell-death events Although inhibition of cell death usually blocks all associated downstream events, a number of experiments have now demonstrated that some aspects of cell-corpse morphology can occur although others are blocked. Paralleling the models of cell-death execution, there may be downstream events that are caspase-activated and those that are mitochondria-activated, or at least caspase-independent. For instance, some downstream events can occur even if caspase function is compromised. During CD2- or staurosporine-induced killing of T lymphocytes, the addition of peptide caspase inhibitors blocked cleavage of PARP and lamins and reduced chromatin condensation; however, loss of mitochondrial transmembrane potential, PS exposure, and cell shrinkage still occurred (Deas et al., 1998). In addition to having an important role in brain development (Kuida et al., 1996; Woo et al., 1998), caspase-3 seems to be especially important for some of the downstream events of death in cells in which its function is not absolutely required for death (Porter and Janicke, 1999). There is a correlation between lack of caspase-3 (in knockout cells or the cell line MCF-7) and lack of DNA degradation, cell shrinkage and plasma membrane blebbing 27 (Janicke et al., 1998; Woo et al., 1998; Zheng et al., 1998); PS externalization does occur in these cells. However, a putative specific inhibitor of caspase-3 (DMQD-CHO) blocked chromatin condensation, DNA fragmentation, apoptotic body formation and PS externalization in response to killing by Fas (Hirata et al., 1998). Conversely, altered mitochondrial function has been associated with a block in plasma membrane changes during apoptosis. The mitochondrial inhibitors antimycin A and oligomycin blocked PS exposure and increased membrane permeability, but not activation of caspase-3, PARP cleavage and DNA fragmentation (Zhuang et al., 1998). Reduction in the mitochondrial transmembrane potential also was blocked by these inhibitors. It is not clear why these inhibitors should prevent aspects of cell death, since they also eliminate mitochondrial function. Release of factors from the mitochondria, such as cytochrome c, AIF and Ca", was not monitored in this study. Defects in downstream events can affect cell killing Failure to induce certain of the downstream events of apoptosis can affect cell killing. Nuclear lamins are cleaved by caspases during programmed cell death (Lazebnik et al., 1995). Rao et al. (1996) introduced lamins with a mutated caspase cleavage site into mammalian cells and examined the kinetics and morphology of cell death. The onset of deaths of these cells was delayed and when cells died, nuclear structural changes of cell death were altered: chromatin condensation, nuclear shrinkage and DNA degradation did not occur initially. However, apoptotic bodies, with pycnotic nuclei, were eventually formed. These observations suggested that cleavage of components of the lamina promotes the nuclear events of apoptosis, possibly by allowing entrance of degradation factors into the nucleus. In Drosophila dcp-1 mutants, which have defects in the apoptotic death of nurse cells, lamin Dm0 is not degraded; in these cells, nuclei remain intact, as assayed by persistence of a nuclearlocalized B-gal marker in dcp-1 but not wild-type nurse cells (McCall and Steller, 1998). Cell-corpse engulfment Cell corpses are clearedfrom living tissues by engulfment After a cell dies by programmed cell death, it is rapidly engulfed, either by professional phagocytes or by neighboring cells. This removal of the dead cell may prevent lysis and damage to the surrounding tissue, and also enable the recycling of 28 components of the dead cell (discussed in Ren and Savill, 1998). The engulfment process involves two steps: signaling from the cell corpse to phagocytic cells and phagocytosis, surrounding the cell corpse by membranes of the phagocytic cell. From work in mammalian systems and Drosophila,several distinct interactions have been implicated in corpse-phagocyte signaling. However, in no case is both the cell-corpse ligand and the engulfing-cell receptor definitively known. C. elegans mutants defining six genes, ced-1, -2, -5, -6, -7 and -10, important for cellcorpse engulfment have been identified. None of the engulfment mutations leads to the persistence of the full complement of cell corpses, although at least some of these mutations are likely to be null (Liu and Hengartner, 1998; Wu and Horvitz, 1998a, b). The six genes appear to fall into two functional groups (ced-1, -6 and -7; ced-2, -5 and -10): double mutants between genes of a single group have no more persistent cell corpses than the stronger single mutant, and double mutants between genes in two groups have somewhat more corpses than in either single mutant (Ellis et al., 1991a). Still, in no case is engulfment completely blocked, suggesting that multiple signals are important and that not all signals have been identified by mutation. It is not clear whether these mutants are blocked in the recognition or phagocytosis step of engulfment, since EM analysis of the mutants showed that there was no extension of pseudopodia from dying cells around the cell corpses examined (Ellis et al., 1991a). Cell-corpse engulfment: possible recognition mechanisms Phosphatidylserine presentation at the plasma membrane may serve as a cellcorpse marker. Phagocytosis of apoptotic lymphocytes can be inhibited by PScontaining liposomes (Fadok et al., 1992a). The mitochondrial inhibitors antimycin A and oligomycin, which prevented PS exposure on dying cells, also blocked the engulfment of those cells (Zhuang et al., 1998). Several engulfing-cell receptors have been suggested to function in PS recognition: Class B scavenger receptors (CD36, SRB1), CD68 (oxidized LDL receptor) and annexins (reviewed by Fadok et al., 1998). A number of additional experiments have implicated scavenger receptors in cellcorpse recognition. Knockout of the Class A scavenger receptor results in reduced binding of apoptotic thymocytes to SRA -/- macrophages in vitro (Platt et al., 1996; Terpstra et al., 1997). Expression of CD36, a Class B receptor, conferred phagocytic ability to a normally non-competent cell line (Ren et al., 1995). The Drosophila gene 29 croquemort (crq) encodes a protein with similarity to CD36 and may be involved in recognizing a signal(s) from the dying cell (Franc et al., 1996). Point mutations in crq have not been described, but animals homozygous for a deficiency including the crq locus are engulfment-defective. Interestingly, the expression of crq is induced by programmed cell deaths: crq is not expressed in embryos in which cell death is blocked, and expression increases in response to treatments such as ionizing radiation that increase the number of cell deaths in the embryo (Franc et al., 1996). Thus, it is possible that in Drosophilasignals from dying cells induce surrounding cells to become engulfment-competent. However, these data also suggest that Crq is not the only receptor for signals from dying cells. u133 integrin may be another receptor on the engulfing cell that recognizes the dying cell or mediates phagocytosis. Blocking integrin function with RGDS peptide or antibodies specific for the integrin subunits decreases engulfment (Savill et al., 1990; Fadok et al., 1992a). Macrophages that are blocked by RGDS or anti-OV0 3 are not blocked by PS liposomes, suggesting that thrombospondin does not bind to apoptotic cells via PS and that the PS receptor and 0v 3 represent distinct signaling systems (Fadok et al., 1992a). While the signal from the cell corpse is not known, thrombospondin has been shown to be involved in bridging this interaction between the dying and engulfing cell (Savill et al., 1989). The C. elegans gene ced-7 encodes an ABC-transporter protein (ATP-binding cassette) (Wu and Horvitz, 1998a). Different ABC transporters function to transport a variety of substrates into or out of the cell. The substrate of CED-7 is not known, and furthermore the sequence of CED-7 is not particularly similar to any ABC-transporter protein with a known substrate. Some ABC transporters have lipid-translocating activity (e.g., Ruetz and Gros, 1994; van Helvoort et al., 1996), so it is possible that CED7 functions in PS exposure on the outer leaflet of the plasma membrane. It is also possible that CED-7 transports a soluble signal across the plasma membrane that is recognized by engulfing cells. Curiously, mosaic analysis of ced-7 indicates that it may be required within both the dying and engulfing cells (Wu and Horvitz, 1998a). A mammalian ABC transporter, ABC1, has been implicated in the engulfment of apoptotic cells (Luciani and Chimini, 1996) and it can function as an anion transporter when expressed in Xenopus oocytes (Becq et al., 1997). 30 Thus, in C. elegans and in other species, multiple signals from the dying cell may contribute to cell-corpse recognition. It is possible that multiple signals might be required to unambiguously mark a cell corpse. It is also likely that different cell types might present different signals. Cell-corpse engulfment: phagocytosis After the recognition step of engulfment, signals must be transduced within the engulfing cell for phagocytosis to proceed. The process by which an engulfing cell extends pseudopodia and embraces the cell corpse must involve cytoskeletal rearrangements and membrane fusion. The C. elegans gene ced-5 may be involved in cytoskeletal rearrangements, since it encodes a protein similar to human DOCK180 and DrosophilaMyoblast city (mbc) (Wu and Horvitz, 1998b). DOCK180 was identified as a protein that binds to Crk (Hasegawa et al., 1996). Crk is involved in transducing signals from integrin receptors to the cytoskeleton, and DOCK180 may mediate these cytoskeletal rearrangements by activating Raci (Kiyokawa et al., 1998). In addition, mbc alleles have been isolated as suppressors of the consequences of Rac overexpression in the Drosophilaeye (Nolan et al., 1998). The C. elegans gene ced-6 encodes an adaptor protein with a phosphotyrosine-binding domain and thus also may be involved in signaling (Liu and Hengartner, 1998). Consistent with their proposed roles in signal transduction in the engulfing cell, ced-6 and ced-5 both appear to function in engulfing cells (Liu and Hengartner, 1998; Wu and Horvitz, 1998b). How do upstream events in programmed cell death trigger the engulfment of a dying cell? It is possible that caspase cleavage generates a molecule that is used for signaling to other cells, inactivates the flippase normally responsible for maintaining plasma membrane asymmetry and/or modifies a cell-surface receptor to modulate the cell corpse's binding properties. Engulfment and cell killing During development of the mammalian eye, the pupillary membrane, a network of capillaries, is formed and then regresses. This remodeling of the eye occurs by programmed cell death (Lang et al., 1994) and is associated with the presence of macrophages during the regression period. Ablation of macrophages by expression of diphtheria toxin under the control of a macrophage-specific promoter resulted in 31 persistence of the pupillary membrane until later developmental stages (Lang and Bishop, 1993). Furthermore, injection of toxic liposomes into the developing rat eye prevented regression of the pupillary membrane and replacement of macrophages rescued this phenotype (Diez-Roux and Lang, 1997). These data suggest that this cell death in the developing eye is induced by macrophages. In C. elegans, the death of one cell is triggered by or in some way requires the function of a neighboring engulfing cell (Sulston et al., 1980). Migration of the male linker cell is required for the outgrowth of the gonad; when outgrowth is completed, death of the linker cell creates an opening for the passage of sperm. In contrast to laser ablations of the neighbors of other cells that die in C. elegans, which did not block programmed cell deaths (Sulston and White, 1980), ablation of a neighbor prevented the death of the linker cell. That the apparent "murdering" cell is also the engulfing cell may be coincidental; i.e., the cell-killing signal may be distinct from engulfment signaling. Engulfment-mutant males are mating-competent, suggesting that death of the linker cell does occur reliably in these animals. Certain mutations in the C. elegans genes lin-24 and lin-33 lead to the deaths of vulval precursor cells and thus result in an egg-laying defect (Ferguson et al., 1987). These deaths do not resemble other C. elegans programmed deaths in appearance, and they are not suppressed by ced-3(lf), ced-4(lf) and ced-9(gf) mutations. However, the Egl phenotype is suppressed by mutations in the engulfment genes ced-2, ced-5 and ced-10, suggesting that the cell deaths are suppressed as well (S. C. Kim, personal communication). In the wild type, engulfment might promote the death of lin-24- and lin-33-affected cells by disposing of the cells quickly as they start to die, and if engulfment is delayed, the cells be able to recover. This model does not explain why mutations in some but not all engulfment genes suppress the lin-24 and lin-33 phenotype. Perhaps lin-24- and lin-33- mediated deaths do not trigger engulfment by programmed cell death-specific mechanisms and therefore are not affected by mutations in genes that function specifically in programmed cell death. How ced-1, -6 and -7 function in the engulfment process is not known, but these genes might be involved in signaling between the dying cell and the engulfing cell, and by comparison, ced-2, -5 and -10 might function in less-specific engulfment processes. 32 Conclusions Programmed cell death is a conserved feature of metazoan development and homeostasis. Important regulators of cell killing have been identified, and these regulators appear to be conserved evolutionarily. Some progress has been made in understanding how the downstream events that define apoptosis might occur, but it remains to be seen how cell killing triggers most of these morphological changes. In some cases, different aspects of the downstream events have been uncoupled from one another, suggesting a possible dichotomy between caspase-dependent and mitochondria-dependent events. However, which caspase substrates represent key regulators of downstream events, and for which downstream events are the mitochondria actually required? Are there other, unidentified regulators of the downstream events? Finally, why are these downstream events so highly-conserved and what is their role in the cell killing process? 33 Acknowledgments I thank Mark Metzstein for critical comments concerning this chapter. I also thank Brad Hersh, Peter Reddien, Zheng Zhou and the other members of the Horvitz Lab PCD community for many helpful discussions of the cell-death literature. 34 References Abrams, J. M., White, K., Fessler, L. I., and Steller, H. (1993). Programmed cell death during Drosophilaembryogenesis. Development 117, 29-43. Adams, J. M., and Cory, S. (1998). The Bcl-2 Protein Family: Arbiters of Cell Survival. Science 281, 322-326. Baker, K. P., Baron, W. F., Henzel, W. J., and Spencer, S. A. (1998). Molecular cloning and characterization of human and murine DNase II. Gene 215, 281-289. Barry, M. A., and Eastman, A. (1992). Endonuclease activation during apoptosis: the role of cytosolic Ca 2 + and pH. Biochem. Biophys. Res. Commun. 186, 782-789. Barry, M. A., and Eastman, A. (1993). Identification of deoxyribonuclease II as an endonuclease involved in apoptosis. Arch. Biochem. Biophys. 300, 440-450. Basse, F., Stout, J. G., Sims, P. J., and Wiedmer, T. (1996). Isolation of an erythrocyte membrane protein that mediates Ca 2 +-dependent transbilayer movement of phospholipid. J. Biol. Chem. 271, 17205-17210. Becq, F., Hamon, Y., Bajetto, A., Gola, M., Verrier, B., and Chimini, G. (1997). ABC1, an ATP binding cassette transporter required for phagocytosis of apoptotic cells, generates a regulated anion flux after expression in Xenopus laevis oocytes. J. Biol. Chem. 272, 2695-2699. Bevers, E. M., Comfurius, P., VanRijn, J. L. M. L., Hemker, H. C., and Zwaal, R. F. A. (1982). Generation of prothrombin-converting activity and the exposure of phosphatidylserine at the outer surface of platelets. Eur. J. Biochem. 122, 429-436. Brancolini, C., Lazarevic, D., Rodriguez, J., and Schneider, C. (1997). Dismantling cellcell contacts during apoptosis is coupled to a caspase-dependent proteolytic cleavage of beta-catenin. J. Cell Biol. 139, 759-771. Bump, N. J., Hackett, M., Hugunin, M., Seshagiri, S., Brady, K., Chen, P., Ferenz, C., Franklin, S., Ghayur, T., Li, P., and et al. (1995). Inhibition of ICE family proteases by baculovirus antiapoptotic protein p35. Science 269, 1885-1888. Cai, J., Yang, J., and Jones, D. P. (1998). Mitochondrial control of apoptosis: the role of cytochrome c. Biochim. Biophys. Acta 1366, 139-149. Cecconi, F., Alvarez-Bolado, G., Meyer, B. I., Roth, K. A., and Gruss, P. (1998). Apafi (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 94, 727-737. Chauhan, D., Pandey, P., Ogata, A., Teoh, G., Krett, N., Halgren, R., Rosen, S., Kufe, D., Kharbanda, S., and Anderson, K. (1997). Cytochrome c-dependent and -independent induction of apoptosis in multiple myeloma cells. J. Biol. Chem. 272, 29995-29997. 35 Chen, P., Nordstrom, W., Gish, B., and Abrams, gene in Drosophila.Genes Dev. 10, 1773-1782. J. M. (1996). grim, a novel cell death Conradt, B., and Horvitz, H. R. (1998). The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell 93, 519-529. Deas, 0., Dumont, C., MacFarlane, M., Rouleau, M., Hebib, C., Harper, F., Hirsch, F., Charpentier, B., Cohen, G. M., and Senik, A. (1998). Caspase-independent cell death induced by anti-CD2 or staurosporine in activated human peripheral T lymphocytes. Immunol. 161, 3375-3383. J. Diez-Roux, G., and Lang, R. A. (1997). Macrophages induce apoptosis in normal cells in vivo. Development 124, 3633-3638. Ellis, H. M., and Horvitz, H. R. (1986). Genetic control of programmed cell death in the nematode C. elegans. Cell 44, 817-829. Ellis, R. E., Jacobson, D. M., and Horvitz, H. R. (1991a). Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditiselegans. Genetics 129, 79-94. Ellis, R. E., Yuan, J. Y., and Horvitz, H. R. (1991b). Mechanisms and functions of cell death. Annu. Rev. Cell Biol. 7, 663-698. Enari, M., Sakahira, H., Yokoyama, H., Okawa, K., Iwamatsu, A., and Nagata, S. (1998). A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391, 43-50. Fadok, V. A., Bratton, D. L., Frasch, D. L., Warner, M. L., and Henson, P. M. (1998). The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ. 5, 551-562. Fadok, V. A., Savill, J. S., Haslett, C., Bratton, D. L., Doherty, D. E., Campbell, P. A., and Henson, P. M. (1992a). Different populations of macrophages use either the vitronectin receptor or the phosphatidylserine receptor to recognize and remove apoptotic cells. J. Immunol. 149, 4029-4035. Fadok, V. A., Voelker, D. R., Campbell, P. A., Cohen, J. J., Bratton, D. L., and Henson, P. M. (1992b). Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207-2216. Ferguson, E. L., Sternberg, P. W., and Horvitz, H. R. (1987). A genetic pathway for the specification of the vulval cell lineages of Caenorhabditiselegans. Nature 326, 259-267. Fesus, L., Thomazy, V., Autuori, F., Ceru, M. P., Tarcsa, E., and Piacentini, M. (1989). Apoptotic hepatocytes become insoluble in detergents and chaotropic agents as a result of transglutaminase action. FEBS Lett. 245, 150-154. 36 Franc, N. C., Dimarcq, J. L., Lagueux, M., Hoffmann, J., and Ezekowitz, R. A. (1996). Croquemort, a novel Drosophilahemocyte/macrophage receptor that recognizes apoptotic cells. Immunity 4,431-443. Gaido, M. L., and Cidlowski, J. A. (1991). Identification, purification, and characterization of a calcium-dependent endonuclease (NUC18) from apoptotic rat thymocytes. NUC18 is not histone H2B. J. Biol. Chem. 266, 18580-18585. Gavrieli, Y., Sherman, Y., and Ben-Sasson, S. A. (1992). Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 119, 493-501. Geng, Y. J., Azuma, T., Tang, J. X., Hartwig, J. H., Muszynski, M., Wu, Q., Libby, P., and Kwiatkowski, D. J. (1998). Caspase-3-induced gelsolin fragmentation contributes to actin cytoskeletal collapse, nucleolysis, and apoptosis of vascular smooth muscle cells exposed to proinflammatory cytokines. Eur J Cell Biol 77, 294-302. Gentile, V., Thomazy, V., Piacentini, M., Fesus, L., and Davies, P. J. A. (1992). Expression of Tissue Transglutaminase in Balb-C 3T3 Fibroblasts: Effects of Cellular Morphology and Adhesion. J. Cell Biol. 119, 463-474. Gottlieb, R. A., Giesing, H. A., Engler, R. L., and Babior, B. M. (1995a). The acid deoxyribonuclease of neutrophils: a possible participant in apoptosis-associated genome destruction. Blood 86, 2414-2418. Gottlieb, R. A., Giesing, H. A., Zhu, J. Y., Engler, R. L., and Babior, B. M. (1995b). Cell acidification in apoptosis: granulocyte colony-stimulating factor delays programmed cell death in neutrophils by up-regulating the vacuolar H(+)-ATPase. Proc. Natl. Acad. Sci. U. S. A. 92, 5965-5968. Gottlieb, R. A., Nordberg, J., Skowronski, E., and Babior, B. M. (1996). Apoptosis induced in Jurkat cells by several agents is preceded by intracellular acidification. Proc. Natl. Acad. Sci. U. S. A. 93, 654-658. Green, D. R., and Reed, J. C. (1998). Mitochondria and apoptosis. Science 281, 1309-1312. Grether, M. E., Abrams, J. M., Agapite, J., White, K., and Steller, H. (1995). The head involution defective gene of Drosophilamelanogasterfunctions in programmed cell death. Genes Dev. 9, 1694-1708. Gumienny, T. L., Lambie, E., Hartwieg, E., Horvitz, H. R., and Hengartner, M. 0. (1999). Genetic control of programmed cell death in the Caenorhabditiselegans hermaphrodite germline. Development 126, 1011-1022. Halenbeck, R., MacDonald, H., Roulston, A., Chen, T. T., Conroy, L., and Williams, L. T. (1998). CPAN, a human nuclease regulated by the caspase-sensitive inhibitor DFF45. Curr. Biol. 8, 537-540. 37 Hampton, M. B., Vanags, D. M., Porn-Ares, M. I., and Orrenius, S. (1996). Involvement of extracellular calcium in phosphatidylserine exposure during apoptosis. FEBS Lett. 399, 277-282. Hasegawa, H., Kiyokawa, E., Tanaka, S., Nagashima, K., Gotoh, N., Shibuya, M., Kurata, T., and Matsuda, M. (1996). DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol. Cell Biol. 16, 1770-1776. Hay, B. A., Wolff, T., and Rubin, G. M. (1994). Expression of baculovirus p35 prevents cell death in Drosophila.Development 120, 2121-2129. Hedgecock, E. M., Sulston, J. E., and Thomson, J. N. (1983). Mutations affecting programmed cell deaths in the nematode Caenorhabditiselegans. Science 220, 1277-1279. Hendzel, M. J., Nishioka, W. K., Raymond, Y., Allis, C. D., Bazett-Jones, D. P., and Th'ng, J. P. (1998). Chromatin condensation is not associated with apoptosis. J. Biol. Chem. 273, 24470-24478. Hengartner, M. 0., Ellis, R. E., and Horvitz, H. R. (1992). Caenorhabditiselegans gene ced9 protects cells from programmed cell death. Nature 356, 494-499. Hengartner, M. 0., and Horvitz, H. R. (1994). C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell 76, 665-676. Hirata, H., Takahashi, A., Kobayashi, S., Yonehara, S., Sawai, H., Okazaki, T., Yamamoto, K., and Sasada, M. (1998). Caspases are activated in a branched protease cascade and control distinct downstream processes in Fas-induced apoptosis. J. Exp. Med. 187, 587-600. Hughes, F. M., and Cidlowski, J. A. (1994). Apoptotic DNA degradation: evidence for novel enzymes. Cell Death Differ. 1, 11-17. Hughes, F. M., Jr., Evans-Storms, R. B., and Cidlowski, J. A. (1998). Evidence that noncaspase proteases are required for chromatin degradation during apoptosis. Cell Death Differ. 5, 1017-1027. Jacobson, M. D., Weil, M., and Raff, M. C. (1997). Programmed cell death in animal development. Cell 88, 347-354. Janicke, R. U., Sprengart, M. L., Wati, M. R., and Porter, A. G. (1998). Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 273, 9357-9360. Jiang, S., Chow, S. C., Nicotera, P., and Orrenius, S. (1994). Intracellular Ca 2 + signals activate apoptosis in thymocytes: studies using the Ca 2 +-ATPase inhibitor thapsigargin. Exp. Cell Res. 212, 84-92. 38 Kaiser, N., and Edelman, I. S. (1977). Calcium dependence of glucocorticoid-induced lymphocytolysis. Proc. Natl. Acad. Sci. U. S. A. 74, 638-642. Kaiser, N., and Edelman, I. S. (1978). Calcium dependence of ionophore A23187induced lymphocyte cytotoxicity. Cancer Res. 38, 3599-3603. Kawahara, A., Ohsawa, Y., Matsumura, H., Uchiyama, Y., and Nagata, S. (1998). Caspase-independent cell killing by Fas-associated protein with death domain. J. Cell Biol. 143, 1353-1360. Kerr, J. F. R., Wyllie, A. H., and Currie, A. R. (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer. 26, 239257. Kidd, V. J. (1998). Proteolytic Activities that Mediate Apoptosis. Ann Rev Physiol 60, 533-573. Kimble, J., and Hirsh, D. (1979). The postembryonic cell lineages of the hermaphrodite and male gonads in Caenorhabditiselegans. Dev. Biol. 70, 396-417. Kiyokawa, E., Hashimoto, Y., Kobayashi, S., Sugimura, H., Kurata, T., and Matsuda, M. (1998). Activation of Raci by a Crk SH3-binding protein, DOCK180. Genes Dev. 12, 3331-3336. Kluck, R. M., Bossy-Wetzel, E., Green, D. R., and Newmeyer, D. D. (1997). The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 275, 1132-1136. Knudson, C. M., and Korsmeyer, S. J. (1997). Bcl-2 and Bax function independently to regulate cell death. Nature Genet 16, 358-363. Kothakota, S., Azuma, T., Reinhard, C., Klippel, A., Tang, J., Chu, K., McGarry, T. J., Kirschner, M. W., Koths, K., Kwiatkowski, D. J., and Williams, L. T. (1997). Caspase-3generated fragment of gelsolin: effector of morphological change in apoptosis. Science 278, 294-298. Krieser, R. J., and Eastman, A. (1998). The cloning and expression of human deoxyribonuclease II. A possible role in apoptosis. J. Biol. Chem. 273, 30909-30914. Kroemer, G., Dallaporta, B., and Resche-Rigon, M. (1998). The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 60, 619-642. Kuida, K., Haydar, T. F., Kuan, C. Y., Gu, Y., Taya, C., Karasuyama, H., Su, M. S., Rakic, P., and Flavell, R. A. (1998). Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 94, 325-337. Kuida, K., Zheng, T. S., Na, S., Kuan, C., Yang, D., Karasuyama, H., Rakic, P., and Flavell, R. A. (1996). Decreased apoptosis in the brain and premature lethality in CPP32deficient mice. Nature 384, 368-372. 39 Kyprianou, N., English, H. F., and Isaacs, J. T. (1988). Activation of a Ca 2 +-Mg 2 +dependent endonuclease as an early event in castration-induced prostatic cell death. Prostate 13, 103-117. Lang, R., Lustig, M., Francois, F., Sellinger, M., and Plesken, H. (1994). Apoptosis during macrophage-dependent ocular tissue remodelling. Development 120, 3395-3403. Lang, R. A., and Bishop, J. M. (1993). Macrophages are required for cell death and tissue remodeling in the developing mouse eye. Cell 74,453-462. Lazebnik, Y. A., Takahashi, A., Moir, R. D., Goldman, R. D., Poirier, G. G., Kaufmann, S. H., and Earnshaw, W. C. (1995). Studies of the lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc. Natl. Acad. Sci. U. S. A. 92, 9042-9046. Lee, N., MacDonald, H., Reinhard, C., Halenbeck, R., Roulston, A., Shi, T., and Williams, L. T. (1997). Activation of hPAK65 by caspase cleavage induces some of the morphological and biochemical changes of apoptosis. Proc. Natl. Acad. Sci. U. S. A. 94, 13642-13647. Levkau, B., Herren, B., Koyama, H., Ross, R., and Raines, E. W. (1998). Caspasemediated cleavage of focal adhesion kinase pp125FAK and disassembly of focal adhesions in human endothelial cell apoptosis. J. Exp. Med. 187, 579-586. Li, J., and Eastman, A. (1995). Apoptosis in an interleukin-2-dependent cytotoxic T lymphocyte cell line is associated with intracellular acidification. Role of the Na+/H+antiport. J. Biol. Chem. 270, 3203-3211. Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S. M., Ahmad, M., Alnemri, E. S., and Wang, X. (1997). Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479-489. Liu, Q. A., and Hengartner, M. 0. (1998). Candidate adaptor protein CED-6 promotes the engulfment of apoptotic cells in C. elegans. Cell 93, 961-972. Liu, Q. Y., Pandey, S., Singh, R. K., Lin, W., Ribecco, M., Borowy-Borowski, H., Smith, B., LeBlanc, J., Walker, P. R., and Sikorska, M. (1998). DNase Y: A Rat DNase I-like gene Coding for a Constitutively Expressed Chromatin-Bound Endonuclease. Biochem 37, 10134-10143. Liu, X., Kim, C. N., Yang, J., Jemmerson, R., and Wang, X. (1996). Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147-157. Liu, X., Zou, H., Slaughter, C., and Wang, X. (1997). DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 89, 175-184. 40 Long, X., Crow, M. T., Sollott, S. J., O'Neill, L., Menees, D. S., Hipolito, M., Boluyt, M. 0., Asai, T., and Lakatta, E. G. (1998). Enhanced Expression of p53 and Apoptosis Induced by Blockade of the Vacuolar Proton ATPase in Cardiomyocytes. J. Clin. Invest. 101, 1453-1461. Luciani, M. F., and Chimini, G. (1996). The ATP binding cassette transporter ABC1, is required for the engulfment of corpses generated by apoptotic cell death. EMBO J. 15, 226-235. Madi, A., Punyiczki, M., di Rao, M., Piacentini, M., and Fesus, L. (1998). Biochemical characterization and localization of transglutaminase in wild-type and cell-death mutants of the nematode Caenorhabditiselegans. Eur. J. Biochem. 253, 583-590. Martin, D. A., Siegel, R. M., Zheng, L., and Lenardo, M. J. (1998). Membrane oligomerization and cleavage activates the caspase-8 (FLICE/MACHalphal) death signal. J. Biol. Chem. 273, 4345-4349. Martin, S. J., Lennon, S. V., Bonham, A. M., and Cotter, T. G. (1990). Induction of apoptosis (programmed cell death) in human leukemic HL-60 cells by inhibition of RNA or protein synthesis. J. Immunol. 145, 1859-1867. Martin, S. J., Reutelingsperger, C. P. M., McGahon, A. J., Rader, J. A., van Schie, R. C. A. A., LaFace, D. M., and Green, D. R. (1995). Early Redistribution of Plasma Membrane Phosphatidylserine Is a General Feature of Apoptosis Regardless of the Initiating Stimulus: Inhibition by Overexpression of Bcl-2 and Abl. J. Exp. Med. 182, 1545-1556. McCall, K., and Steller, H. (1998). Requirement for DCP-1 caspase during Drosophila oogenesis. Science 279, 230-234. Meisenholder, G. W., Martin, S. J., Green, D. R., Nordberg, J., Babior, B. M., and Gottlieb, R. A. (1996). Events in apoptosis. Acidification is downstream of protease activation and BCL-2 protection. J. Biol. Chem. 271, 16260-16262. Melino, G., and Piacentini, M. (1998). 'Tissue' transglutaminase in cell death: a downstream or a multifunctional upstream effector? FEBS Lett. 430, 59-63. Metzstein, M. M., Stanfield, G. M., and Horvitz, H. R. (1998). Genetics of programmed cell death in C. elegans: Past, present and future. Trends Genet. 14, 410-416. Minn, A. J., Velez, P., Schendel, S. L., Liang, H., Muchmore, S. W., Fesik, S. W., Fill, M., and Thompson, C. B. (1997). Bcl-xL forms an ion channel in synthetic lipid membranes. Nature 385, 353-357. Montague, J. W., Gaido, M. L., Frye, C., and Cidlowski, J. A. (1994). A calciumdependent nuclease from apoptotic rat thymocytes is homologous with cyclophilin. Recombinant cyclophilins A, B, and C have nuclease activity. J. Biol. Chem. 269, 1887718880. 41 Montague, J. W., Hughes, F. M., Jr., and Cidlowski, J. A. (1997). Native recombinant cyclophilins A, B, and C degrade DNA independently of peptidylprolyl cis-transisomerase activity. Potential roles of cyclophilins in apoptosis. J. Biol. Chem. 272, 66776684. Nagata, S. (1996). Fas-induced apoptosis, and diseases caused by its abnormality. Genes Cells 1, 873-879. Nikonova, L. V., Beletsky, I. P., and Umansky, S. R. (1993). Properties of some nuclear nucleases of rat thymocytes and their changes in radiation-induced apoptosis. Eur. J. Biochem. 215, 893-901. Nolan, K. M., Barrett, K., Lu, Y., Hu, K. Q., Vincent, S., and Settleman, J. (1998). Myoblast city, the Drosophilahomolog of DOCK180/CED-5, is required in a Rac signaling pathway utilized for multiple developmental processes. Genes Dev. 12, 33373342. Oberhammer, F., Fritsch, G., Schmied, M., Bavelka, M., Printz, D., Purchio, T., Lassmann, H., and Schulte-Hermann, R. (1993a). Condensation of the chromatin at the membrane of an apoptotic nucleus is not associated with activation of an endonuclease. J. Cell Sci. 104, 317-326. Oberhammer, F., Wilson, J. W., Dive, C., Morris, I. D., Hickman, J. A., Wakeling, A. E., Walker, P. R., and Sikorska, M. (1993b). Apoptotic death in epithelial cells: cleavage of DNA to 300 and/or 50 kb fragments prior to or in the absence of internucleosomal fragmentation. EMBO J. 12, 3679-3684. Page, A. P., MacNiven, K., and Hengartner, M. 0. (1996). Cloning and biochemical characterization of the cyclophilin homologues from the free-living nematode Caenorhabditiselegans. Biochem. J. 317, 179-185. Peitsch, M. C., Hesterkamp, T., Polzar, B., Mannherz, H. G., and Tschopp, J. (1992). Functional characterisation of serum DNase I in MRL-lpr/lpr mice. Biochem. Biophys. Res. Commun. 186, 739-745. Peitsch, M. C., Polzar, B., Stephan, H., Crompton, T., MacDonald, H. R., Mannherz, H. G., and Tschopp, J. (1993). Characterization of the endogenous deoxyribonuclease involved in nuclear DNA degradation during apoptosis (programmed cell death). EMBO J. 12, 371-377. Pettmann, B., and Henderson, C. E. (1998). Neuronal cell death. Neuron 20, 633-647. Platt, N., Suzuki, H., Kurihara, Y., Kodama, T., and Gordon, S. (1996). Role for the class A macrophage scavenger receptor in the phagocytosis of apoptotic thymocytes in vitro. Proc. Natl. Acad. Sci. U. S. A. 93, 12456-12460. Porter, A. G., and Janicke, R. U. (1999). Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 6, 99-104. 42 Rao, L., Perez, D., and White, E. (1996). Lamin proteolysis facilitates nuclear events during apoptosis. J. Cell Biol. 135, 1441-1455. Reed, J. C., Jurgensmeier, J. M., and Matsuyama, S. (1998). Bcl-2 family proteins and mitochondria. Biochim. Biophys. Acta 1366, 127-137. Ren, Y., and Savill, Differ. 5, 563-568. J. (1998). Apoptosis: The importance of being eaten. Cell Death Ren, Y., Silverstein, R. L., Allen, J., and Savill, J. (1995). CD36 gene transfer confers capacity for phagocytosis of cells undergoing apoptosis. J. Exp. Med. 181, 1857-1862. Robertson, A. M. G., and Thomson, J. N. (1982). Morphology of programmed cell death in the ventral nerve cord of Caenorhabditiselegans larvae.J. Embryol. exp. Morphol. 67, 89-100. Rudel, T., and Bokoch, G. M. (1997). Membrane and morphological changes in apoptotic cells regulated by caspase-mediated activation of PAK2. Science 276, 1571-1574. Ruetz, S., and Gros, P. (1994). Phosphatidylcholine translocase: a physiological role for the mdr2 gene. Cell 77, 1071-1081. Sakahira, H., Enari, M., and Nagata, S. (1998). Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 391, 96-99. Samejima, K., Tone, S., Kottke, T. J., Enari, M., Sakahira, H., Cooke, C. A., Durrieu, F., Martins, L. M., Nagata, S., Kaufmann, S. H., and Earnshaw, W. C. (1998). Transition from caspase-dependent to caspase-independent mechanisms at the onset of apoptotic execution. J. Cell Biol. 143, 225-239. Saunders, J. W., Gasseling, M. T., and Saunders, L. C. (1962). Cellular death in morphogenesis of the avian wing. Dev. Biol. 139, 169-185. Savill, J., Dransfield, I., Hogg, N., and Haslett, C. (1990). Vitronectin receptor-mediated phagocytosis of cells undergoing apoptosis. Nature 343, 170-173. Savill, J. S., Henson, P. M., and Haslett, C. (1989). Phagocytosis of aged human neutrophils by macrophages is mediated by a novel "charge-sensitive" recognition mechanism. J. Clin. Invest. 84, 1518-1527. Schendel, S. L., Xie, Z., Montal, M. 0., Matsuyama, S., Montal, M., and Reed, J. C. (1997). Channel formation by antiapoptotic protein Bcl-2. Proc. Natl. Acad. Sci. U. S. A. 94, 5113-5118. Schlesinger, P. H., Gross, A., Yin, X. M., Yamamoto, K., Saito, M., Waksman, G., and Korsmeyer, S. J. (1997). Comparison of the ion channel characteristics of proapoptotic BAX and antiapoptotic BCL-2. Proc. Natl. Acad. Sci. U. S. A. 94, 11357-11362. 43 Shaham, S. (1998). Identification of multiple Caenorhabditiselegans caspases and their potential roles in proteolytic cascades. J. Biol. Chem. 273, 35109-35117. Shaham, S., and Horvitz, H. R. (1996). Developing Caenorhabditiselegans neurons may contain both cell-death protective and killer activities. Genes Dev. 10, 578-591. Shibasaki, F., and McKeon, F. (1995). Calcineurin functions in Ca 2 +-activated cell death in mammalian cells. J. Cell Biol. 131, 735-743. Slee, E. A., Harte, M. T., Kluck, R. M., Wolf, B. B., Casiano, C. A., Newmeyer, D. D., Wang, H., Reed, J. C., Nicholson, D. W., Alnemri, E. S., Green, D. R., and Martin, S. J. (1999). Ordering the Cytochrome c-initiated Caspase Cascade: Hierarchical Activation of Caspases-2, -3, -6, -7, -8, and -10 in a Caspase-9-dependent Manner. J. Cell Biol. 144, 281-292. Stroh, C., and Schulze-Osthoff, K. (1998). Death by a thousand cuts: an ever increasing list of caspase substrates. Cell Death Differ. 5, 997-1000. Sugimoto, A., Friesen, P. D., and Rothman, J. H. (1994). Baculovirus p35 prevents developmentally programmed cell death and rescues a ced-9 mutant in the nematode Caenorhabditiselegans. EMBO J. 13, 2023-2028. Sulston, J. E., Albertson, D. G., and Thomson, J. N. (1980). The Caenorhabditiselegans male: postembryonic development of nongonadal structures. Dev. Biol. 78, 542-576. Sulston, J. E., and Horvitz, H. R. (1977). Post-embryonic cell lineages of the nematode Caenorhabditiselegans. Dev. Biol. 56, 110-156. Sulston, J. E., Schierenberg, E., White, J. G., and Thomson, J. N. (1983). The embryonic cell lineage of the nematode Caenorhabditiselegans. Dev. Biol. 100, 64-119. Sulston, J. E., and White, J. G. (1980). Regulation and cell autonomy during postembryonic development of Caenorhabditiselegans. Dev. Biol. 78, 577-597. Susin, S. A., Lorenzo, H. K., Zamzami, N., Marzo, I., Snow, B. E., Brothers, G. M., Mangion, J., Jacotot, E., Costantini, P., Loeffler, M., Larochette, N., Goodlett, D. R., Aebersold, R., Siderovski, D. P., Penninger, J. M., and Kroemer, G. (1999). Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397, 441-446. Susin, S. A., Zamzami, N., Castedo, M., Daugas, E., Wang, H. G., Geley, S., Fassy, F., Reed, J. C., and Kroemer, G. (1997). The central executioner of apoptosis: multiple connections between protease activation and mitochondria in Fas/APO-1/CD95- and ceramide-induced apoptosis. J. Exp. Med. 186, 25-37. Susin, S. A., Zamzami, N., Castedo, M., Hirsch, T., Marchetti, P., Macho, A., Daugas, E., Geuskens, M., and Kroemer, G. (1996). Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J. Exp. Med. 184, 1331-1341. 44 Susin, S. A., Zamzami, N., and Kroemer, G. (1998). Mitochondria as regulators of apoptosis: doubt no more. Biochim. Biophys. Acta 1366, 151-165. Terpstra, V., Kondratenko, N., and Steinberg, D. (1997). Macrophages lacking scavenger receptor A show a decrease in binding and uptake of acetylated low-density lipoprotein and of apoptotic thymocytes, but not of oxidatively damaged red blood cells. Proc. Natl. Acad. Sci. U. S. A. 94, 8127-8131. Thompson, C. B. (1995). Apoptosis in the pathogenesis and treatment of disease. Science 267, 1456-1462. Thornberry, N. A., and Lazebnik, Y. (1998). Caspases: enemies within. Science 281, 13121316. Toh, S. Y., Wang, X., and Li, P. (1998). Identification of the nuclear factor HMG2 as an activator for DFF nuclease activity. Biochem. Biophys. Res. Commun. 250, 598-601. Torriglia, A., Chaudun, E., Chany-Fournier, F., Jeanny, J. C., Courtois, Y., and Counis, M. F. (1995). Involvement of DNase II in nuclear degeneration during lens cell differentiation. J. Biol. Chem. 270, 28579-28585. Torriglia, A., Perani, P., Brossas, J. Y., Chaudun, E., Treton, J., Courtois, Y., and Counis, M. F. (1998). L-DNase II, a molecule that links proteases and endonucleases in apoptosis, derives from the ubiquitous serpin leukocyte elastase inhibitor. Mol. Cell Biol. 18, 3612-3619. Urbano, A., McCaffrey, R., and Foss, F. (1998). Isolation and characterization of NUC70, a cytoplasmic, hematopoietic apoptotic endonuclease. J. Biol. Chem. 273, 34820-34827. Van de Craen, M., Van Loo, G., Pype, S., Van Criekinge, W., Van den Brande, I., Molemans, F., Fiers, W., Declerq, W., and Vandenabeele, P. (1998). Identification of a new caspase homologue: caspase-14. Cell Death Differ. 5, 838-846. van Helvoort, A., Smith, A. J., Sprong, H., Fritzsche, I., Schinkel, A. H., Borst, P., and van Meer, G. (1996). MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell 87, 507517. Varkey, J., Chen, P., Jemmerson, R., and Abrams, J. M. (1999). Altered Cytochrome c Display Precedes Apoptotic Cell Death in Drosophila.J. Cell Biol. 144, 701-710. Vaux, D. L., and Korsmeyer, S. J. (1999). Cell death in development. Cell 96, 245-254. Vermes, I., Haanen, C., Steffens-Nakken, H., and Reutelingsperger, C. (1995). A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods 184, 3951. 45 Wakabayashi, S., Shigekawa, M., and Pouyssegur, J. (1997). Molecular Physiology of Vertebrate Na+/H+ Exchangers. Physiol Rev 77, 51-74. Wang, H. G., Pathan, N., Ethell, I. M., Krajewski, S., Yamaguchi, Y., Shibasaki, F., McKeon, F., Bobo, T., Franke, T. F., and Reed, J. C. (1999). Ca 2 +-Induced Apoptosis Through Calcineurin Dephosphorylation of BAD. Science 284, 339-343. White, K., Grether, M. E., Abrams, J. M., Young, L., Farrell, K., and Steller, H. (1994). Genetic control of programmed cell death in Drosophila.Science 264, 677-683. Wolf, C. M., and Eastman, A. (1999). Intracellular Acidification during Apoptosis Can Occur in the Absence of a Nucleus. Biochem. Biophys. Res. Commun. 254, 821-827. Woo, M., Hakem, R., Soengas, M. S., Duncan, G. S., Shahinian, A., Kagi, D., Hakem, A., McCurrach, M., Khoo, W., Kaufman, S. A., Senaldi, G., Howard, T., Lowe, S. W., and Mak, T. W. (1998). Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev. 12, 806-819. Wu, Y. C., and Horvitz, H. R. (1998a). The C. elegans cell corpse engulfment gene ced-7 encodes a protein similar to ABC transporters. Cell 93, 951-960. Wu, Y. C., and Horvitz, H. R. (1998b). C. elegans phagocytosis and cell-migration protein CED-5 is similar to human DOCK180. Nature 392, 501-504. Wyllie, A. H. (1980). Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature 284, 555-556. Wyllie, A. H., Kerr, J. F. R., and Currie, A. R. (1980). Cell death: the significance of apoptosis. Int. Rev. Cytol. 68, 251-306. Wyllie, A. H., Morris, R. G., Smith, A. L., and Dunlop, D. (1984). Chromatin cleavage in apoptosis: association with condensed chromatin morphology and dependence on macromolecular synthesis. J Pathol 142, 67-77. Yang, J., Liu, X., Bhalla, K., Kim, C. N., Ibrado, A. M., Cai, J., Peng, T. I., Jones, D. P., and Wang, X. (1997). Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275, 1129-1132. Yasuda, T., Takeshita, H., lida, R., Nakajima, T., Hosomi, 0., Nakashima, Y., and Kishi, K. (1998). Molecular cloning of the cDNA encoding human deoxyribonuclease II. J. Biol. Chem. 273, 2610-2616. Yoshida, H., Kong, Y. Y., Yoshida, R., Elia, A. J., Hakem, A., Hakem, R., Penninger, J. M., and Mak, T. W. (1998). Apafi is required for mitochondrial pathways of apoptosis and brain development. Cell 94, 739-750. 46 Yuan, J., Shaham, S., Ledoux, S., Ellis, H. M., and Horvitz, H. R. (1993). The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 betaconverting enzyme. Cell 75, 641-652. Yuan, J. Y., and Horvitz, H. R. (1990). The Caenorhabditiselegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev. Biol. 138, 33-41. Zhang, C., Robertson, M. J., and Schlossman, S. F. (1995). A triplet of nuclease proteins (NP42-50) is activated in human Jurkat cells undergoing apoptosis. Cell Immunol. 165, 161-167. Zhang, J., Liu, X., Scherer, D. C., van Kaer, L., Wang, X., and Xu, M. (1998). Resistance to DNA fragmentation and chromatin condensation in mice lacking the DNA fragmentation factor 45. Proc. Natl. Acad. Sci. U. S. A. 95, 12480-12485. Zheng, T. S., Schlosser, S. F., Dao, T., Hingorani, R., Crispe, I. N., Boyer, J. L., and Flavell, R. A. (1998). Caspase-3 controls both cytoplasmic and nuclear events associated with Fas-mediated apoptosis in vivo. Proc Natl Acad Sci USA 95, 13618-13623. Zhou, Q., Zhao, J., Stout, J. G., Luhm, R. A., Wiedmer, T., and Sims, P. J. (1997). Molecular cloning of human plasma membrane phospholipid scramblase. A protein mediating transbilayer movement of plasma membrane phospholipids. J. Biol. Chem. 272, 18240-18244. Zhuang, J., Ren, Y., Snowden, R. T., Zhu, H., Gogvadze, V., Savill, J. S., and Cohen, G. M. (1998). Dissociation of phagocyte recognition of cells undergoing apoptosis from other features of the apoptotic program. J. Biol. Chem. 273, 15628-15632. Zoratti, M., and Szabo, I. (1995). The mitochondrial permeability transition. Biochim. Biophys. Acta 1241, 139-176. Zou, H., Henzel, W. J., Liu, X., Lutschg, A., and Wang, X. (1997). Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90, 405-413. Zwaal, R. F., and Schroit, A. J. (1997). Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood 89, 1121-1132. 47 Chapter 2 The ced-8 gene controls the timing of programmed cell deaths in C. elegans Gillian M. Stanfield & H. Robert Horvitz To be submitted to Cell 48 Summary Loss-of-function mutations in the gene ced-8 lead to the late appearance of cell corpses during embryonic development in C. elegans. We found that these cell corpses result from a late-initiating or slowly-occurring cell-death program. ced-8 functions downstream of or in parallel to the regulatory cell-death gene ced-9 and may function as a cell-death effector downstream of the caspase encoded by the programmed cell death killer gene ced-3. ced-8 encodes a transmembrane protein that appears to be localized to the plasma membrane. The CED-8 protein is similar to human XK, a putative membrane transport protein implicated in McLeod Syndrome, a form of hereditary neuroacanthocytosis. 49 Introduction It is critical for animals to be able to eliminate unwanted cells in a regulated, timely manner in response to developmental and environmental cues. This elimination is accomplished through a tightly-controlled process of programmed cell death. The functions of specific cell-death genes within cells that die are required for their demise, and a number of the key molecules that control this process have been identified and are conserved among animals (reviewed in Vaux and Korsmeyer, 1999). Failure to kill appropriate cells can lead to severe developmental defects (e.g., Kuida et al., 1996; Hakem et al., 1998; Kuida et al., 1998; Varfolomeev et al., 1998). In the Caenorhabditiselegans hermaphrodite, 131 of the 1090 somatic cells generated during development are destined to die (Sulston and Horvitz, 1977; Sulston et al., 1983). The identities of the cells that undergo programmed cell death in C. elegans are essentially invariant from animal to animal. The majority of cell deaths occur early in development during embryonic stages coincident with a period of rapid cell division followed by the initiation of morphogenesis. In genetic screens searching for mutants abnormal in the pattern of cell corpses, C. elegans mutants variously defective in the specification, execution, engulfment or degradation of programmed cell deaths have been identified. Analyses of these mutants have provided a framework for understanding how cells die by programmed cell death, not only in C. elegans but also in other animals (reviewed by Metzstein et al., 1998). The activities of ced-3 (cell death abnormal), which encodes a caspase (Yuan et al., 1993; Xue et al., 1996), and ced-4, which encodes a protein similar to mammalian Apaf-1 (Yuan and Horvitz, 1992; Zou et al., 1997), are required for cells to undergo programmed cell death in C. elegans (Ellis and Horvitz, 1986). Cells destined to survive are protected by the activity of the gene ced-9, which encodes a member of the Bcl-2 family of proteins (Hengartner and Horvitz, 1994b); in dying cells, ced-9 is negatively regulated by the gene egl-1 (egg-laying defective), which encodes a BH3 (Bcl-2 homology 3) domain-containing protein (Conradt and Horvitz, 1998). As a cell dies by programmed cell death, morphological changes in many aspects of its structure are visible (Kerr et al., 1972). These changes define the process of apoptosis. The plasma membrane blebs, and the cell becomes detached from its neighbors. As observed using electron microscopy (EM), the cytoplasm condenses and becomes darkly staining (electron-dense) while organelles remain grossly intact. The DNA becomes condensed at 50 the nuclear periphery and is degraded by endonuclease activity; cell corpses can be visualized using the TUNEL (terminal transferase-mediated dUTP nick end labeling) technique, which labels DNA nicks in situ (Gavrieli et al., 1992). The dying cell may fragment into smaller membrane-bound bodies. The dying cell is eventually engulfed and degraded within the engulfing cell. By most morphological criteria, cell corpses in C. elegans are similar to cells dying by apoptosis in mammals, although dying cells in C. elegans do not fragment into smaller bodies. As visualized by Nomarski microscopy, C. elegans cell corpses are highly refractile and easily distinguished from their living sisters (Sulston and Horvitz, 1977). These refractile corpses probably correspond to the electron-dense stage seen by EM (Robertson and Thomson, 1982). The DNA of C. elegans cell corpses is degraded (Sulston, 1976), and C. elegans cell corpses can be visualized using TUNEL (Y.-C. Wu, G.M.S. and H.R.H., manuscript in preparation). Although many genes important for controlling the killing step of programmed cell death have been identified, it is not clear how downstream events, such as the morphological changes that occur as cells die and the engulfment and degradation of cell corpses, are effected. Caspases are thought to be key mediators of these downstream events (e.g., Liu et al., 1997; Janicke et al., 1998), but while many in vitro targets of caspase proteolysis have been identified, the functions of these targets in cell death are in general unclear. DNA degradation may be triggered by the caspase-dependent cleavage of a protein that binds to and inhibits a cell-death endonuclease (Enari et al., 1998; Sakahira et al., 1998). Several molecules implicated in the engulfment process have been characterized recently (Franc et al., 1996; Liu and Hengartner, 1998; Wu and Horvitz, 1998a, b). Nonetheless, it is not known how the initiation of programmed cell death leads to the engulfment of a dying cell or whether the triggering of the engulfment process is a direct or indirect result of caspase activity. In general, the role of molecules that may function as caspase substrates or that otherwise act downstream of effector caspases is not understood. For instance, some activities might be needed for aspects of cell killing, while others might be important for degradation. It is also unclear whether any downstream molecules in the cell-death process are activated by caspase-independent mechanisms. The conservation among diverse organisms of both the morphology of dying cells and the molecules implicated in cell killing suggests that aspects of cell killing downstream of the key regulatory molecules are likely to be conserved. For this reason, genetic analysis of 51 programmed cell death in C. elegans, which has identified these regulatory molecules, should also permit the characterization of the events that occur downstream of cell-death initiation and of how these downstream events contribute to the cell-killing process. In this report, we demonstrate that the gene ced-8 is involved in the execution step of programmed cell death in C. elegans and that the function of ced-8 is important for the proper kinetics of programmed cell death during development. We cloned the ced-8 gene and found that it is similar to XK, a putative membrane transport protein from humans (Ho et al., 1994). We suggest that ced-8 functions in the downstream processes of cell killing. Results The appearance of cell corpses is delayed in ced-8 mutant embryos During the approximately 14 hours of C. elegans embryogenesis, cell divisions and cell deaths together result in a total of 558 cells as the developing animal is transformed from a single cell into a vermiform larva capable of movement and feeding (Sulston et al., 1983). Most somatic cells destined to die in C. elegans do so soon after they are generated, and most cell corpses are visible during the developmental period between the bean and 2-fold stages when many cells are dividing and the embryo begins to elongate (Sulston and Horvitz, 1977; Sulston et al., 1983) (Figure 1A). As viewed by Nomarski microscopy, a dying cell becomes highly refractile and distinct from its living neighbors. The refractile disk of the dying cell disappears during the engulfment process, which occurs within an hour of the initiation of the cell's death, and engulfed corpses are not visible at all. The total number of cell corpses visible at any time during development therefore represents a balance between the number of cells that have died up to that time and the number of cell corpses that have been engulfed. ced-8 was identified in a screen for C. elegans mutants containing an increased number of cell corpses at the late embryonic and early larval stages (Ellis et al., 1991). Other mutants identified in this screen are defective in the engulfment of cell corpses by neighboring cells. In engulfment-defective mutants, cell deaths occur at the appropriate time during development but cell corpses persist in an unengulfed state (Hedgecock et al., 1983; Ellis et al., 1991) (Figure 1B). By contrast, we found that in ced-8 mutants the majority of cell corpses did not appear until late in embryogenesis, and the greatest number of cell corpses was visible in late 3-fold embryos (Figure 1C, Table 1). Furthermore, nearly all 52 these cell corpses disappeared by the time of hatching, suggesting that they are indeed rapidly engulfed. We observed a clear shift toward a later appearance of cell corpses than in the wild type for all ced-8 alleles examined with the exception of n3113, which was identified as a suppressor of the lethality conferred by ced-4(n2273) ced-9(n 1653) (Shaham and Horvitz, 1996a; E. Speliotes and H.R.H., personal communication) and was nearly indistinguishable from the wild type in cell-corpse assays (Table 1). To confirm that the appearance of cell corpses is delayed in ced-8 mutants, we examined the effect of a ced-8 mutation on cell corpse number in a ced-7; ced-5 background, in which most cell corpses remain unengulfed (Ellis et al., 1991). We found that most cell corpses in ced-7; ced-5; ced-8 embryos were visible during the 3-fold stage of embryogenesis, just as in the ced-8 single mutants (Figure ID). We saw no more corpses in early ced-7; ced-5; ced-8 embryos than in early ced-8 embryos, consistent with the interpretation that cell corpses appear late, rather than persist, in animals mutant for ced-8. Furthermore, the total number of cell corpses that accumulates by the late 3-fold stage of embryogenesis in ced-7; ced-5; ced-8 animals is similar to the maximum number observed in ced-7; ced-5 animals, consistent with the idea that a delay rather than a reduction in cell killing is caused by mutations in ced-8. ced-8 has a weak effect on cell killing Strong loss-of-function (lf) mutations in ced-3 and ced-4 result in the survival of nearly all cells that normally die in C. elegans (Ellis and Horvitz, 1986). Weak loss-of-function mutations in ced-3 and ced-4 result in the survival of an apparently random subset of those cells that normally die (Hengartner and Horvitz, 1994a; S. Shaham, B. Davies, P. Reddien and H.R.H., manuscript in preparation), and some cell deaths are delayed (M. Hengartner, personal communication). Based on the number of corpses in embryos, ced-8 mutations did not appear to cause many cells to survive ectopically. However, since some mutations in ced-3 and ced-4 can affect the kinetics of cell death, we examined whether any extra cells were present in the anterior pharynx of ced-8 mutant animals. Counts of extra cells in the anterior region of the pharynx, the nematode feeding organ, are a useful measurement of the degree to which cell death is blocked in a ced mutant (Hengartner et al., 1992). For instance, strong mutations in ced-3 or ced-4 result in approximately 13 extra cells in this region of the animal (Table 2). We found that very few if any extra cells were present in ced-8 mutants as compared to strong ced-3 and ced-4 mutants (Tables 1, 2). 53 One possible explanation for the weak nature of the ced-8 phenotype was that the known ced-8 mutations might cause only a partial loss of function of this gene. To test whether strong ced-8(lf) mutations might cause a more pronounced cell-survival phenotype, we scored for extra cells in the pharynges of ced-8(n1891)/yDf2 animals. We found that there was no significant difference in the number of extra cells in ced-8(n1891)/yDf2 animals as compared to the number in ced-8(n1891) homozygotes (Table 1). Furthermore, there was no decrease in viability of embryos from lon-2(e678) ced-8(n1891)/yDf2 mothers as compared to embryos from lon-2(e678)/yDf2 mothers; in both cases, approximately a quarter of embryos failed to hatch (data not shown), as expected if only the yDJ2 homozygous class of progeny was inviable in each strain. These results are consistent with n1891's being a strong loss-of-function or null allele of ced-8. Although mutations in ced-8 alone did not result in the survival of a significant number of cells that normally die, it was possible that ced-8 might enhance the weak cell-survival phenotype that results from partial loss of ced-3 or ced-4 function. We scored for the presence of extra cells in the anterior pharynx of animals mutant both for ced-8 and for the weak ced-3 mutations n2427 or n2438. We found that a ced-8 mutation and a weak ced-3 mutation had a synergistic effect on the number of extra cells in the anterior pharynx (Table 2). We also examined animals mutant for both ced-8 and ced-4(n2273), a weak ced-4 mutation that results in the survival of only a few cells that normally die. ced-4(n2273); ced-8(n1891) animals also contained more extra cells than did either of the single mutants (Table 1, 2). Thus, mutations in ced-8 enhance the cell-survival phenotype of all weak ced-3 and ced-4 mutants tested. Double mutants between ced-8 and the strong alleles ced3(n1040), ced-3(n717) or ced-4(n1162) had no more extra cells than did the ced-3 or ced-4 single mutant animals, suggesting that ced-8 does not function in a cell-death pathway parallel to that defined by ced-3 and ced-4. These data are consistent with a modulatory function for ced-8 in cell killing. ced-8 appears to play a role in cell death that is distinct from those of ced-3 and ced-4: ced-8 is not absolutely required for cell killing, but during development it enhances the efficiency of those deaths that occur. ced-8 acts downstream of or in parallel to ced-9 The gene ced-9 is required for cell survival during development and for fertility: ced-9(lf) mutations result in ectopic cell deaths leading to maternal-effect lethality and zygotic 54 sterility (Hengartner et al., 1992). Mutations in either ced-3 or ced-4 suppress the ced-9(lf) phenotype, indicating that the lethality results from ectopic cell deaths and that ced-9 normally functions to negatively regulate ced-3 and ced-4 activity. To test whether ced-8 acts genetically upstream of ced-9, we determined whether ced-9 function is required for the enhanced cell-survival phenotype of a ced-3(weak); ced-8 double mutant. We found that more extra cells are present in ced-9(n2812); ced-3(n2427); ced-8(n1891) mutant animals than are present in ced-9(n2812); ced-3(n2427) animals (P < 0.0001, paired t test) (Table 3). ced-9(n2812) is likely to be null for ced-9 function (Hengartner and Horvitz, 1994a). Thus, mutations in ced-8 can enhance cell survival even in the absence of all ced-9 activity, indicating that ced-8 acts downstream of or in parallel to ced-9. DNA degradation in ced-8 mutants How can one distinguish whether ced-8 mutants initiate programmed cell death late or rather execute the process of programmed cell death slowly? If programmed cell deaths were initiated late, then the appearance of all markers of cell death should be delayed, but once killing is triggered, the morphological changes that result in a refractile corpse would presumably occur with normal kinetics. However, if programmed cell deaths were initiated at the normal time in ced-8 mutants, then some cell-death markers could appear at the appropriate time even though the progression of the cell to refractility is slowed. To attempt to distinguish between these possible effects if ced-8, we sought additional biochemical markers of cell corpses besides the refractility visible by Nomarski microscopy. We reasoned that if cell death were initiated at the proper time in ced-8 animals, it might be possible to find a cell-death marker that would appear with wild-type kinetics in ced-8 mutants. Degradation of the nuclear DNA of dying cells is a hallmark of apoptosis and can be visualized in situ using TUNEL (TdT-mediated dUTP Nick End Labeling) (Gavrieli et al., 1992 and Y.-C. Wu, G.M.S. and H.R.H., manuscript in preparation). In labeled wildtype C. elegans embryos, a small fraction of dying cells are TUNEL-reactive at any one time; embryos mutant for the gene nuc-1, which is important for DNA degradation (Sulston, 1976; Hevelone and Hartman, 1988), contain many condensed TUNEL-reactive nuclei that appear when cells die and many of these persist throughout embryogenesis (Figure 2A, B; Y.-C. Wu, G.M.S. and H.R.H., manuscript in preparation). Thus, DNA degradation is apparently very rapid during programmed cell death in wild-type C. elegans and DNA degradation is slowed in nuc-1 mutants relative to in the wild type. Furthermore, 55 engulfment per se is not necessary for the generation of TUNEL-reactive DNA in cell corpses (Y.-C. Wu, G.M.S. and H.R.H., manuscript in preparation), suggesting that TUNEL represents a cell-intrinsic marker for cell deaths. We therefore examined whether mutations in ced-8 affect the kinetics of DNA degradation as visualized by TUNEL. In ced-8 embryos, as in wild-type embryos, few TUNEL-positive corpses were present at any given developmental stage (Figure 2C). However, in a ced-8 nuc-1 double mutant strain, the appearance of many TUNEL-positive corpses seemed to be delayed until late in embryogenesis at the time at which many late-appearing refractile cell corpses are visible by Nomarski microscopy. The appearance of TUNEL-reactive corpses in a ced-8 nuc-1 mutant strain is consistent with the idea that ced-8 function is not required for generating TUNEL-reactive nicks in cell-corpse DNA. The late appearance of most TUNEL-positive corpses is consistent with our observations, based on examining refractile corpses, that most cell corpses appear late in embryogenesis in ced-8 mutants. To compare the time course of the appearance of TUNEL-positive corpses with the time course of the appearance of refractile cell corpses, we examined TUNEL-stained ced-8 nuc-1 double mutant animals at early developmental time points during embryogenesis and compared the number of TUNEL-positive cells to the number of refractile corpses visible in a ced-7; ced-5; ced-8 mutant strain at the same developmental stage. The nuc-1 DNA degradation-defective and the ced-7; ced-5 engulfment-defective backgrounds allow a similar number of accumulated cell corpses to be visualized at each developmental stage (Figure 2B vs. Figure 1B), suggesting that these two methods of visualizing corpses have similar sensitivities. We found that in early embryos there were many more corpses in ced-8 nuc-1 embryos as visualized by TUNEL staining than there were refractile corpses in ced-7; ced-5; ced-8 mutant embryos as visualized by Nomarski optics (Figure 2D vs. Figure 1D). That so many TUNEL-positive cells were present at this early stage suggests that although morphological changes causing refractility are delayed in most cell corpses until the 3-fold stage of embryogenesis, DNA degradation is less delayed. This result is consistent with the hypothesis that cell death is triggered at the appropriate developmental time in ced-8 mutants but proceeds to the refractile-corpse stage more slowly than in the wild type and that the biochemical and cytological changes of dying cells that occur nearly synchronously in the wild type are uncoupled from one another in ced-8 mutants. 56 Cloning of ced-8 ced-8 previously had been mapped to LGX to the right of unc-10, near dpy-6 (Ellis et al., 1991). We more precisely mapped ced-8 to the left of dpy-6, which lies to the left of nP81 (Kornfeld et al., 1995). We injected cosmids from the region to the left of nP81 (Figure 3A) into ced-8(n1999) hermaphrodites and found that arrays containing either of the cosmids F54F4 or F37F1 restored cell corpses in early embryos. We localized the region required for rescue to a 4.2 kilobase (kb) fragment from the region of overlap between F54F4 and F37F1 (Figure 3B). We determined the sequence of this DNA fragment and found that it contains a single gene predicted by the program GENEFINDER (L. Hillier and P. Green, personal communication). We identified a ced-8 cDNA (Figure 3C; see Materials and Methods) that rescued the Ced-8 phenotype when expressed in ced-8(n1891) embryos under the control of C. elegans heat-shock promoters (data not shown). To determine whether we had identified the 5' end of this rescuing transcript, we analyzed 5' RACE products from C. elegans embryonic RNA and performed northern analysis. ced-8 transcripts identified by RACE had 41 additional nucleotides of contiguous genomic sequence as compared to the longest cDNAs isolated from phage libraries. A northern blot identified a single band migrating at the approximate size of the putative full-length transcript in RNA from embryos or LI larvae but not in RNA from older animals (Figure 4). We found no evidence of longer transcripts that might result from cis-splicing to upstream sequences or of trans-splicingto C. elegans splice leaders (Krause and Hirsh, 1987). Since the ced-8 transcript has two in-frame stop codons six base pairs (bp) 5' to the putative transcriptional start and there are no splice acceptor sites before these stop codons, it is likely that we have identified the complete ced-8 coding sequence. To confirm that we had identified the correct gene, we determined the sequences of the ced-8 coding region from ced-8 mutants and identified mutations for all nine alleles (Figure 3C). We found that the lesions in the ced-8 alleles are consistent with their causing loss of ced-8 function. Most known ced-8 mutations cause an identical phenotype that is consistent with a strong or complete loss of function. However, the n3113 mutation, which eliminates the splice acceptor for intron 3 and is predicted to result in an in-frame insertion of 16 amino acids, results in a weaker phenotype: very few cell deaths are delayed until late stages of embryogenesis in n3113 embryos (Table 1). 57 Similarity of CED-8 to XK and other proteins The ced-8 cDNA encodes a 458 amino acid protein weakly similar to the product of the human XK gene (Figure 5A) (Ho et al., 1994). CED-8 and XK share 19% identity and 37% similarity overall and have similar hydropathy plots (Figure 5B). Both proteins contain 10 hydrophobic predicted transmembrane-spanning segments, and most sequence similarity between the two proteins lies within these regions. Consistent with these structural predictions for CED-8, a ced-8:gfp (green fluorescent protein) translational fusion, which includes most of the ced-8 coding region and rescued the delayed-death phenotype of ced-8(n1999) embryos (data not shown), was localized to the plasma membrane (Figure 6). Two regions of similarity between CED-8 and XK are shared with a predicted protein from the ascidian Ciona intestinalis,and one of these regions is also shared with a Drosophila EST (Figure 5C). Discussion ced-8 mutations delay the appearance of cell corpses ced-8 was originally identified in a screen for animals defective in the engulfment process of programmed cell death (Ellis et al., 1991). That screen sought to identify mutants based on the presence of visible cell corpses in late embryonic and early larval stages. ced-8 mutants, like other mutants identified in that screen, contain visible cell corpses at these late stages and thus ced-8 was categorized initially as an engulfment gene. However, by examining earlier stages of development, we discovered that corpses were not visible in ced-8 embryos at the stages when corpses normally appear both in the wild type and in engulfment mutants. This observation revealed a primary role for ced-8 in a step of cell death prior to the engulfment of cell corpses. Specifically, ced-8 appears to be involved in controlling the timing of programmed cell death. We note that Ellis et al. (1991) found that as visualized by electron microscopy embryonic corpses in ced-8 animals were not engulfed, while postembryonic corpses in the ventral cord of ced-8 larvae were engulfed. This disparity suggested that ced-8 might function specifically to engulf embryonic cell corpses. We believe that these observations are consistent with our hypothesis that ced-8 mutations affect the timing of programmed cell death. Specifically, since corpses, once they appear in ced-8 mutants, do not appear to persist, they likely are engulfed quickly. It is possible that the embryonic corpses that were 58 not visibly engulfed in ced-8 mutants (Ellis et al., 1991) had just died and engulfment had not yet begun; although visible signs of engulfment can occur during the division that generates a cell doomed to die (Robertson and Thomson, 1982), the engulfment of very few corpses has been analyzed at a resolution that could indicate whether signs of engulfment generally appear so quickly. However, it is also possible that one effect of loss of ced-8 is to slow the engulfment process even after a refractile corpse has appeared. Such an effect might not reflect a direct requirement for ced-8 in the engulfment process; cells dying late might not be engulfed with the same kinetics as cells dying at the normal time during development simply because not all components of the engulfment-recognition system are widely expressed at all stages of development (Wu and Horvitz, 1998b). Clearly, the cellular milieu of a late-dying cell is very different from that of a cell dying at the normal time. We have shown that ced-8 is important in C. elegans to ensure that cell deaths occur on time and that in sensitized genetic backgrounds ced-8 is important to ensure that the proper number of cell deaths occurs. While expression of ced-8 under the control of heat-shock promoters was able to rescue the mutant phenotype of ced-8(n1891) animals, no additional abnormalities were observed: e.g., there was no increase in the number of cell deaths or any loss of viability associated with overexpression of the ced-8 cDNA (data not shown). By contrast, overexpression of ced-3 or ced-4 either under the control of heat-shock promoters or under cell type-specific promoters is able to induce programmed cell death (Shaham and Horvitz, 1996b; S. Shaham and H.R.H., unpublished). Thus, while ced-8 is neither required for programmed cell death nor sufficient to induce cell killing, ced-8 enhances the efficiency of the cell-death program during development. Our data are consistent with ced-8's acting in either of two steps of the cell-death process: initiation (activation of the cell-death machinery) and execution (promotion of cell killing after activation of the core execution genes has already occurred). Function of ced-8 in programmed cell death Programmed cell death in C. elegans is believed to be initiated molecularly by the activation of the core cell-death pathway, consisting of egl-1, ced-9, ced-4 and ced-3 (Metzstein et al., 1998). Morphological changes and other downstream events are thought to be dependent on cleavage by CED-3 of cell-death substrates, although it is not known precisely how the cell-killing process results in the refractility or most other morphological characteristics of a 59 cell corpse. We have found that ced-8, like ced-3 and ced-4, functions downstream of or in parallel to ced-9. Thus, ced-8 could function either to activate ced-3 and/orced-4, i.e., in the initiation of cell killing, or to effect cell killing after cell death has been initiated by ced-3 and ced-4. In the latter case, CED-8 might function as a substrate of CED-3 or by interacting with such a substrate(s). In the former case, ced-8 might act directly on either ced-3 or ced-4 to potentiate their activities and thereby initiate the cell-death process. For instance, CED-8 might function as an adaptor protein between CED-3 and a protein required for the activation of CED-3 enzymatic activity. ced-4 appears to function genetically upstream of ced-3 (Shaham and Horvitz, 1996b), and biochemical studies suggest that CED-4 helps induce the conversion of pro-CED-3 to active CED-3 enzyme (Chinnaiyan et al., 1997; Seshagiri and Miller, 1997; Wu et al., 1997a). When coexpressed with CED-9 in mammalian cells, CED-4 is localized to intracellular membranes and the perinuclear region (Wu et al., 1997b). We observed that a ced-8:gfp fusion is localized to the plasma membrane, suggesting that CED-8 does not function in the CED-4-dependent activation of CED-3. Alternatively, CED-8 might function to localize and thereby activate other proteins that promote cell killing or to sequester and thereby inactivate proteins that inhibit cell killing. A localization role for CED-8 is plausible given the Ced-8 phenotype of delayed death. If ced-8 functions in a rate-enhancing role in the formation of a cell-killing complex, it is reasonable that cell death occurs eventually, albeit more slowly, in its absence. Alternatively, ced-8 might function in some aspect of cell-death execution downstream of ced-3 and ced-4. We have found that the timing of certain downstream cell-death events, the onset of refractility and DNA degradation, are uncoupled in ced-8 cell corpses. Defects in the timing and/or morphology of cell killing may be a common result of compromising events downstream of the central cell-killing pathway. The Ced-8 phenotype is reminiscent of the effect of blocking the cleavage of lamins during apoptosis in mammalian cells (Rao et al., 1996). When lamins with a mutated caspase-cleavage site were introduced into mammalian cells in culture, a delay in death associated with changes in the morphology of dying cells was observed, suggesting that caspase activity in the nucleus facilitates the later events of cell death. How does mutation of ced-8 in a sensitized ced-3 or ced-4 genetic background result in the survival of cells that normally die? It is possible that if a cell death is delayed sufficiently then that cell can lose its competence to die, either because cell-killing 60 protein(s) are no longer present or because cell-protective function(s) are activated. While such defects in cell death have a very minor effect on C. elegans, which is viable even if cell death is essentially completely blocked, it is possible that inefficient cell killing would cause more serious defects in other organisms. Evolutionary conservation of CED-8 CED-8 shares similarity with human XK, the loss of which is implicated in McLeod Syndrome, a hereditary disease defined by abnormalities in erythrocyte Kell antigens (Ho et al., 1994). McLeod erythrocytes also have an acanthocytic (spiculated) appearance, and the McLeod phenotype comprises progressive myopathy and neurodegeneration. The cellular basis of these defects is not understood. We propose, by analogy with the role of ced-8 in C. elegans, that XK may function in apoptosis in humans and that neurodegeneration associated with McLeod Syndrome is a consequence of disruption in apoptosis. The XK protein is associated with the Kell protein in the erythrocyte plasma membrane, and loss of XK leads to the absence of Kell antigens at the cell surface (Khamlichi et al., 1995; Daniels et al., 1996; Russo et al., 1998). The Kell protein is similar to neprilysin-like membrane metallopeptidases, although its substrate is not known (Turner and Tanzawa, 1997). XK has been postulated to function as a transporter (Ho et al., 1994), so one possibility is that XK transports either a substrate or a cleavage product of Kell out of or into the cell. By analogy, it is possible that CED-8 functions in transporting some substrate important for effecting the downstream events of programmed cell death. However, although the two proteins share some sequence similarity and have similar hydropathy profiles, and both are apparently localized to the plasma membrane, the similarity between CED-8 and XK is not extensive. Functional analysis of CED-8, XK and the other related proteins is needed to determine the extent of their functional similarity. One proposed basis for acanthocytic cell morphology is asymmetry between the inner and outer leaflets of the plasma membrane, which could be caused by increased outward translocation of phospholipids (Zwaal et al., 1993; Handin et al., 1995). Exposure of phosphatidylserine on the outer surface of the plasma membrane of dying cells has been implicated in the recognition of cell corpses by engulfing cells (Fadok et al., 1992). CED-8 might function to modulate the lipid content of the plasma membrane, effecting both the 61 refractile characteristic of the cell corpse and its engulfment and thereby coupling the execution of cell death to the morphological changes of the cell corpse. 62 Experimental procedures Nematodes C. elegans were cultured on NGM agar seeded with E. coli strain OP50 as described by Brenner (1974) and were grown at 20'C. Worms used for RNA preparations were grown in liquid culture essentially as described by Wood et al. (1988). All strains were derived from the wild-type Bristol strain N2. All mutations are described in Riddle et al. (1997) unless otherwise noted. A list of the mutations used follows. LG III: ced-7(n1892); ced-9(n2812) (Hengartner and Horvitz, 1994a); qC1(dpy-19(e1259ts) glp-l(q339)) (Epstein and Shakes, 1995); ced-4(n1162, n2273). LG IV: ced-3(n717); ced-3(n2427, n2438) (Hengartner and Horvitz, 1994a); ced-3(n2452) (S. Shaham, B. Davies, P. Reddien and H.R.H., unpublished results); ced-5(n1812). LGX: nuc-1(e1392); unc-10(e102); lon-2(e678); xol-1(y9); dpy-6(e14); yDf2 (L. Miller and B. Meyer, personal communication); szT1(lon-2(e678)). The ced-8 alleles n1891, n1999, n2090, and n2093 are described in Ellis et al. (1991). ced-8(n3113) and ced-8(n3115) were isolated as suppressors of the lethality and sterility caused by ced-4(n2273) ced-9(n1653) (Shaham and Horvitz, 1996a and E. Speliotes and H.R.H., unpublished results), ced-8(n3244) and ced-8(n3245) were isolated as containing late cell corpses (Z. Zhou and H.R.H., unpublished results) and ced-8(n3313) was isolated as an enhancer of the extra-cell phenotype of ced-3(n2427) (P. Reddien and H.R.H., unpublished results). Genetic mapping From the progeny of unc-l0(e102) xol-1(y9) dpy-6/ced-8(n1891) heterozygotes, we selected 56 Unc non-Dpy recombinants and found that 53 segregated Ced-8 progeny, indicating that ced-8 lies between unc-10 and dpy-6, very close to dpy-6 and therefore left of the physical marker nP81 (Kornfeld et al., 1995). Cell Death Assays The number of extra cells in the anterior pharynx of L4 animals was scored using Nomarski microscopy as described by Hengartner et al. (1992). TUNEL experiments were performed as described by Wu et al. (manuscript in preparation; see Chapter 4). After washing, fixed samples were incubated in 1 pg/ml DAPI (4'-6-diamidino-2-phenylindole dihydrochloride) 63 in PBST for 10 minutes. Animals were staged using the pattern of DAPI-stained nuclei to identify developmental landmarks. ced-8 dosage experiments We crossed lon-2 or lon-2 ced-8 males to szTl/+; unc-32(e189); yDf2/szTl hermaphrodites. Non-Lon cross progeny were scored for extra cells in the pharynx. To assay viability, several of these non-Lon hermaphrodites were picked to a plate and allowed to lay eggs for 2 to 3 hours. The number of eggs was counted immediately after removal of the adult worms and then counted again after at least 16 hours, and the number of worms reaching at least the L3 stage was counted 2 days later. Rescue experiments We performed injections as described by Mello et al. (1991) using the coinjection markers pRF4 at 50-75 ng/pl and egl-5:gfp (A. Chisholm and H.R.H., unpublished results) at 50-75 ng/pl, and we selected transgenic lines using the Roller phenotype. We injected cosmids at 20 ng/pl each into ced-8(n1999) hermaphrodites. We examined 3-fold embryos that expressed the egl-5:gfp marker for the presence of cell corpses in the head region; embryos with fewer than three corpses in the head were scored as rescued. We confirmed that the absence of cell corpses was a consequence of rescue of the Ced-8 phenotype rather than a block in cell death by examining bean- and comma-stage embryos from candidate lines for cell corpses; embryos with more than six corpses in the head at this stage were scored as rescued. We injected heat-shock constructs at 50 ng/pl each into ced-8(n1891) hermaphrodites. For heat-shock experiments, gravid Rol hermaphrodites were allowed to lay eggs for 1.5-2 hours, the adults were removed from the plates and the plates were incubated at 33'C for one hour. The number of corpses was counted when embryos reached either the bean and comma stages or the 3-fold stage of development. Non-heat-shocked control animals were studied in parallel and treated ideltically, except that the plates were placed at 20'C rather than 33'C for one hour. Molecular biology Standard molecular biology procedures were followed (Sambrook et al., 1989). The sequences of primers used for determining DNA sequences, for PCR amplification from 64 ced-8 mutants, and for 5'-RACE are available upon request. We determined sequence from pGS23 by a shotgun protocol using an ABI 373A sequencer. CED-8 corresponds to the predicted gene F08F1.5 (Wilson et al., 1994), except that the splice donor predicted by GENEFINDER for intron six of F08F1.5 is incorrect. We isolated RNA from developmentally synchronized nematodes by standard methods and performed poly(A) selection using Fast Track (Invitrogen). 5'-RACE reactions were performed on embryonic RNA using the 5' RACE system (Gibco). cDNA analysis To identify ced-8 cDNAs, a 3.5 kb HpaI-SphI fragment of pGS23 was used to probe an embryonic-stage cDNA library in gtlI (Okkema and Fire, 1994) and a mixed-stage library in XZAP (Barstead and Waterston, 1989). We found two classes of alternatively spliced cDNA clones in addition to a clone containing introns 1 and 3, which were spliced out of all other isolated cDNAs. The two classes of alternatively spliced cDNA clones differed in two ways: first, the splice donor for the second intron differed between the two clones; second, the Type II cDNA was shorter at the 5' end. The Type I splicing pattern was present in five independent clones, while the Type II splicing pattern was present in only a single independent clone. The Type I cDNA is predicted to encode a 458 amino acid protein. The longest ORF from the Type II cDNA can encode a protein corresponding to the 363 C-terminal amino acids of the Type I ORF. Plasmids pGS35 (Type I) and pGS33 (Type II) were constructed by digesting phage DNA with BsiWI and ligating the insert fragment into pSL1190 (Brosius, 1989) digested with BsiWI. To determine whether either or both of the two alternatively-spliced transcripts encode(s) functional CED-8 protein, we placed the longest ORF from each of the two transcripts under the control of C. elegans heat-shock promoters (Fire et al., 1990) and assayed the ability of these heat shock-cDNA constructs to rescue the Ced-8 phenotype of ced-8(n1891) embryos. Only the Type I ORF rescued, suggesting that the Type II transcript does not encode a functional ced-8 gene. To assess further the possibility that the Type II transcript might be relevant for either the function or regulation of ced-8, we cloned and determined the sequence of the ced-8 region from the closely related nematode C. briggsae. We obtained clones containing genomic C. briggsaeced-8 by probing a XCharon4 library (T. Snutch and D. Baillie, personal communication) and a fosmid library (Genome Systems; fosmid clones were provided by 65 the C. elegans Genome Sequencing Consortium). We found that the Type I splice donor sequence is conserved from C. elegans to C. briggsae,but the Type II splice donor site is absent in C. briggsae. We also found that the majority of the predicted amino acid sequence of C. briggsaeced-8 is well conserved as compared to that of C. elegans Type I, and in particular, the coding potential in the region spliced out of the ced-8 message in the event of a Type II splice donor choice is identical between C. elegans and C. briggsae. Our findings that the Type II long ORF does not appear to encode a functional protein and that the splice site necessary to generate a Type II transcript are not conserved between C. elegans and C. briggsae are consistent with the idea that the Type II cDNA identified in the library is not functional. This cDNA may represent an aberrant transcript that is not normally produced. As compared to the Type I splice donor, the Type II splice donor sequence is more similar to the C. elegans consensus acceptor site (Epstein and Shakes, 1995), so perhaps this donor sometimes may be used aberrantly by splicing machinery even though an unproductive transcript is produced as a result. We consider the Type I cDNA, referred to in the text as the ced-8 cDNA, to be the functional ced-8 product. 66 Acknowledgments We thank B. James for determining DNA sequences; A. Coulson, J. Schein and the C. elegans Genome Sequencing Consortium for DNA clones; B. Barstead and P. Okkema for C. elegans cDNA libraries; T. Snutch and D. Baillie for the C. briggsae genomic library; A. Chisholm for egl-5:gfp; and E. Speliotes, Z. Zhou and P. Reddien for ced-8 alleles. We also thank B. Hersh, M. Metzstein and R. Ranganathan for helpful comments concerning the manuscript. G.M.S. was supported by a Howard Hughes Medical Institute Predoctoral Fellowship. H.R.H. is an Investigator of the Howard Hughes Medical Institute. 67 References Barstead, R. J., and Waterston, R. H. (1989). The basal component of the nematode densebody is vinculin. J. Biol. Chem. 264, 10177-10185. Brenner, S. (1974). The genetics of Caenorhabditiselegans. Genetics 77, 71-94. Brosius, J. (1989). Superpolylinkers in cloning and expression vectors. DNA 8, 759-777. Chinnaiyan, A. M., Chaudhary, D., O'Rourke, K., Koonin, E. V., and Dixit, V. M. (1997). Role of CED-4 in the activation of CED-3. Nature 388, 728-729. Conradt, B., and Horvitz, H. R. (1998). The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell 93, 519-529. Daniels, G. L., Weinauer, F., Stone, C., Ho, M., Green, C. A., Jahn-Jochem, H., Offner, R., and Monaco, A. P. (1996). A combination of the effects of rare genotypes at the XK and KEL blood group loci results in absence of Kell system antigens from the red blood cells. Blood 88, 4045-4050. Ellis, H. M., and Horvitz, H. R. (1986). Genetic control of programmed cell death in the nematode C. elegans. Cell 44, 817-829. Ellis, R. E., Jacobson, D. M., and Horvitz, H. R. (1991). Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditiselegans. Genetics 129, 79-94. Enari, M., Sakahira, H., Yokoyama, H., Okawa, K., Iwamatsu, A., and Nagata, S. (1998). A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391, 43-50. Epstein, H. F., and Shakes, D. C. (1995). Caenorhabditiselegans: Modern Biological Analysis of an Organism. In Methods in Cell Biology, L. Wilson and P. Matsudaira, eds. (San Diego, California: Academic Press, Inc.). Fadok, V. A., Voelker, D. R., Campbell, P. A., Cohen, J. J., Bratton, D. L., and Henson, P. M. (1992). Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207-2216. Fire, A., Harrison, S. W., and Dixon, D. (1990). A modular set of lacZ fusion vectors for studying gene expression in Caenorhabditiselegans. Gene 93, 189-198. Franc, N. C., Dimarcq, J. L., Lagueux, M., Hoffmann, J., and Ezekowitz, R. A. (1996). Croquemort, a novel Drosophilahemocyte/macrophage receptor that recognizes apoptotic cells. Immunity 4, 431-443. Gavrieli, Y., Sherman, Y., and Ben-Sasson, S. A. (1992). Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 119, 493-501. 68 Hakem, R., Hakem, A., Duncan, G. S., Henderson, J. T., Woo, M., Soengas, M. S., Elia, A., de la Pompa, J. L., Kagi, D., Khoo, W., Potter, J., Yoshida, R., Kaufman, S. A., Lowe, S. W., Penninger, J. M., and Mak, T. W. (1998). Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 94, 339-352. Handin, R. I., Lux, S. E., and Stossel, T. P. (1995). Blood: Principles and Practice of Hematology (Philadelphia, Pennsylvania: J. B. Lippincott). Hedgecock, E. M., Sulston, J. E., and Thomson, J. N. (1983). Mutations affecting programmed cell deaths in the nematode Caenorhabditiselegans. Science 220, 1277-1279. Hengartner, M. 0., Ellis, R. E., and Horvitz, H. R. (1992). Caenorhabditiselegans gene ced-9 protects cells from programmed cell death. Nature 356, 494-499. Hengartner, M. 0., and Horvitz, H. R. (1994a). Activation of C. elegans cell death protein CED-9 by an amino-acid substitution in a domain conserved in Bcl-2. Nature 369, 318-320. Hengartner, M. 0., and Horvitz, H. R. (1994b). C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell 76, 665-676. Hevelone, J., and Hartman, P. S. (1988). An endonuclease from Caenorhabditiselegans: partial purification and characterization. Biochem. Genet. 26, 447-461. Ho, M., Chelly, J., Carter, N., Danek, A., Crocker, P., and Monaco, A. P. (1994). Isolation of the gene for McLeod syndrome that encodes a novel membrane transport protein. Cell 77, 869-880. Janicke, R. U., Sprengart, M. L., Wati, M. R., and Porter, A. G. (1998). Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 273, 9357-9360. Kerr, J. F. R., Wyllie, A. H., and Currie, A. R. (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer. 26, 239-257. Khamlichi, S., Bailly, P., Blanchard, D., Goossens, D., Cartron, J. P., and Bertrand, 0. (1995). Purification and partial characterization of the erythrocyte Kx protein deficient in McLeod patients. Eur. J. Biochem. 228, 931-934. Kornfeld, K., Hom, D. B., and Horvitz, H. R. (1995). The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell 83, 903-913. Krause, M., and Hirsh, D. (1987). A trans-spliced leader sequence on actin mRNA in C. elegans. Cell 49, 753-761. Kuida, K., Haydar, T. F., Kuan, C. Y., Gu, Y., Taya, C., Karasuyama, H., Su, M. S., Rakic, P., and Flavell, R. A. (1998). Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 94, 325-337. 69 Kuida, K., Zheng, T. S., Na, S., Kuan, C., Yang, D., Karasuyama, H., Rakic, P., and Flavell, R. A. (1996). Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 384, 368-372. Kyte, J., and Doolittle, R. F. (1982). A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105-132. Liu, Q. A., and Hengartner, M. 0. (1998). Candidate adaptor protein CED-6 promotes the engulfment of apoptotic cells in C. elegans. Cell 93, 961-972. Liu, X., Zou, H., Slaughter, C., and Wang, X. (1997). DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 89, 175-184. Mello, C. C., Kramer, J. M., Stinchcomb, D., and Ambros, V. (1991). Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959-3970. Metzstein, M. M., Stanfield, G. M., and Horvitz, H. R. (1998). Genetics of programmed cell death in C. elegans: Past, present and future. Trends Genet. 14, 410-416. Okkema, P. G., and Fire, A. (1994). The Caenorhabditiselegans NK-2 class homeoprotein CEH-22 is involved in combinatorial activation of gene expression in pharyngeal muscle. Development 120, 2175-2186. Rao, L., Perez, D., and White, E. (1996). Lamin proteolysis facilitates nuclear events during apoptosis. J. Cell Biol. 135, 1441-1455. Riddle, D. L., Blumenthal, T., Meyer, B. J., and Priess, J. R. (1997). C. elegans II (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press). Robertson, A. M. G., and Thomson, J. N. (1982). Morphology of programmed cell death in the ventral nerve cord of Caenorhabditiselegans larvae. J. Embryol. exp. Morphol. 67, 89-100. Russo, D., Redman, C., and Lee, S. (1998). Association of XK and Kell blood group proteins. J. Biol. Chem. 273, 13950-13956. Sakahira, H., Enari, M., and Nagata, S. (1998). Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 391, 96-99. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press). Seshagiri, S., and Miller, L. K. (1997). Caenorhabditiselegans CED-4 stimulates CED-3 processing and CED-3-induced apoptosis. Curr. Biol. 7,455-460. Shaham, S., and Horvitz, H. R. (1996a). An alternatively spliced C. elegans ced-4 RNA encodes a novel cell death inhibitor. Cell 86, 201-208. 70 Shaham, S., and Horvitz, H. R. (1996b). Developing Caenorhabditiselegans neurons may contain both cell-death protective and killer activities. Genes Dev. 10, 578-591. Sulston, J. E. (1976). Post-embryonic development in the ventral cord of Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 275, 287-297. Sulston, J. E., and Horvitz, H. R. (1977). Post-embryonic cell lineages of the nematode Caenorhabditiselegans. Dev. Biol. 56, 110-156. Sulston, J. E., Schierenberg, E., White, J. G., and Thomson, J. N. (1983). The embryonic cell lineage of the nematode Caenorhabditiselegans. Dev. Biol. 100, 64-119. Turner, A. J., and Tanzawa, K. (1997). Mammalian membrane metallopeptidases: NEP, ECE, KELL, and PEX. FASEB J. 11, 355-364. Varfolomeev, E. E., Schuchmann, M., Luria, V., Chiannilkulchai, N., Beckmann, J. S., Mett, I. L., Rebrikov, D., Brodianski, V. M., Kemper, 0. C., Kollet, 0., Lapidot, T., Soffer, D., Sobe, T., Avraham, K. B., Goncharov, T., Holtmann, H., Lonai, P., and Wallach, D. (1998). Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apol, and DR3 and is lethal prenatally. Immunity 9, 267-276. Vaux, D. L., and Korsmeyer, S. J. (1999). Cell death in development. Cell 96, 245-254. Wilson, R., Ainscough, R., Anderson, K., Baynes, C., Berks, M., Bonfield, J., Burton, J., Connell, M., Copsey, T., Cooper, J., et al. (1994). 2.2 Mb of contiguous nucleotide sequence from chromosome III of C. elegans. Nature 368, 32-38. Wood, W. B., and the Community of C. elegans Researchers (1988). The Nematode Caenorhabditiselegans (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press). Wu, D., Wallen, H. D., Inohara, N., and Nunez, G. (1997a). Interaction and regulation of the Caenorhabditiselegans death protease CED-3 by CED-4 and CED-9. J. Biol. Chem. 272, 2144921454. Wu, D., Wallen, H. D., and Nunez, G. (1997b). Interaction and regulation of subcellular localization of CED-4 by CED-9. Science 275, 1126-1129. Wu, Y. C., and Horvitz, H. R. (1998a). The C. elegans cell corpse engulfment gene ced-7 encodes a protein similar to ABC transporters. Cell 93, 951-960. Wu, Y. C., and Horvitz, H. R. (1998b). C. elegans phagocytosis and cell-migration protein CED-5 is similar to human DOCK180. Nature 392, 501-504. Xue, D., Shaham, S., and Horvitz, H. R. (1996). The Caenorhabditiselegans cell-death protein CED-3 is a cysteine protease with substrate specificities similar to those of the human CPP32 protease. Genes Dev. 10, 1073-1083. 71 Yuan, J., and Horvitz, H. R. (19;2). The Caenorhabditiselegans cell death gene ced-4 encodes a novel protein and is expressed during the period of extensive programmed cell death. Development 116, 309-320. Yuan, J., Shaham, S., Ledoux, S., Ellis, H. M., and Horvitz, H. R. (1993). The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell 75, 641-652. Zou, H., Henzel, W. J., Liu, X., Lutschg, A., and Wang, X. (1997). Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90, 405-413. Zwaal, R. F., Comfurius, P., and Bevers, E. M. (1993). Mechanism and function of changes in membrane-phospholipid asymmetry in platelets and erythrocytes. Biochem. Soc. Trans. 21, 248-253. 72 Table 1. ced-8 alleles Genotype wild type No. corpses earlya 10.6 ± 0.6 No. corpses lateb 2.3 ± 0.5 No. extra cellsC 0.04 Range extra cellsd 0.03 0-1 ced-8(n 1891) 0.9 ± 0.2 19.5 ± 1.1 0.7 0.1 0-3 ced-8(n 1999) 0.4 ± 0.1 12.0 ± 0.5 0.4 0.09 0-2 ced-8(n2090) 1.8 ± 0.3 19.0 1.2 0.8 0.1 0-3 ced-8(n2093) 1.8 ± 0.3 19.5 ± 1.5 0.6 0.1 0-3 ced-8(n3113) 9.7 ± 0.6 2.1 ± 0.4 0.1 0.06 0-1 ced-8(n3115) 1.9 ±0.2 16.7 ± 0.8 0.4 0.1 0-2 ced-8(n3244) 0.7 ± 0.2 19.5 ± 1.1 0.7 0.2 0-2 ced-8(n3245) 1.4 ± 0.2 17.1 ± 1.0 0.9 0.2 0-4 ced-8(n3313) ND ND 0.8 0.2 0-3 Ion-2(e678) ced-8(n 189 1)/yDf2 ND ND 0.8 0.2 0-3 Ion-2(e678)/yDf2 ND ND 0.1 0.07 0-1 + a average number of corpses in the head of bean and comma-stage embryos ± sem. ND, not determined. b average number of corpses in the head of 3-fold stage embryos ± sem. c average number of extra cells in the anterior pharynx of L3 and L4 hermaphrodites ± sem. d range in number of extra cells observed in the anterior pharynx. Table 2. ced-8 enhances weak ced-3 and ced-4 alleles Genotype ced-3(n2427) ced-8(n 1891) ced-8(n 1999) ced-3(n2427); ced-8(n1891) C ced-3(n2427); ced-8(n 1999) ced-3(n2438) ced-3(n2438); ced-8(n 1891) ced-3(n2438); ced-8(n1999) ced-3(n 1040) ced-3(n1040); ced-8(n1891) ced-3(n 1040); ced-8(n 1999) ced-3(n717) ced-3(n717); ced-8(n1891)c ced-4(n2273) ced-4(n2273); ced-8(n 1891) ced-4(n2273); ced-8(n1999) ced-4(n 1162) ced-4(n 1162); ced-8(n 1891) C No. extra cellsa 1.0 ± 0.1 0.7 ± 0.1 0.4 ± 0.09 5.2 ± 0.3 4.8 ± 0.3 2.0 ± 0.3 6.5 ± 0.3 5.0 ± 0.3 11.5 0.3 10.3 0.5 0.4 11.1 12.8 0.4 12.3 0.3 3.9 ± 0.3 7.1 ± 0.4 7.0 ± 0.4 13.1 ± 0.3 13.3 ± 0.4 nb Range 42 73 50 30 29 28 32 26 33 23 24 30 30 10 10 10 31 28 0-4 0-3 0-2 3-10 2-8 0-7 3-11 3-8 8-14 6-16 8-15 8-18d 9-15 3-6 5-9 5-8 10-16 9-16 a Average number of cells in the anterior pharynx of L3 and L4 hermaphrodites ± s.e.m. b n, number of animals examined. c These strains contained the mutation Ion-2(e678), which does not affect cell survival. d Although 16 cells die in the anterior region of the pharynx, 18 extra cells were observed in one animal of this genotype. These cells likely originated in the posterior bulb of the pharynx, since the pharynx is separated from other cells by a basement membrane. Extra cells are often slightly misplaced in strong ced mutants. Table 3. ced-8 functions downstream of or in parallel to ced-9 No. extra cellsa nb ced-9(n2812); ced-3(n2427) 7.1 ± 0.4 18 ced-9(n2812); ced-3(n2427); unc- 1O(e 102) ced-8(n 1891) 10.0 ±0.3 21 Genotype aAverage number of extra cells in the anterior pharynx in L3 and L4 hermaphrodites ± s.e.m. bn, number of animals examined. Figures Figure 1. Delayed appearance of programmed cell deaths in ced-8 animals. (A) N2 (B) ced-7(nl892); ced-5(nl812) (C) ced-8(nl891) (D) ced-7(nl892); ced-5(nl812); ced-8(n1891). The y axis represents the average number of corpses visible in the heads of embryos. Stages of embryos examined: bean and comma-stage embryos (B/C); 1.5-fold and 2-fold embryos (1.5/2X); early 3-fold embryos that are 3.5 times the length of the egg but do not yet have a well-developed pharynx (3XE); late 3-fold embryos, which have a welldeveloped pharynx and grinder (3XL); and early LI larvae with four cells in the gonad (Li). Error bars indicate one standard error of the mean (sem). At least 14 embryos of each stage were examined using Nomarski microscopy. 76 Figure 1 Rn A 60- N2 55 50- 50 - 45- 45- 40 20 40 - - ced-7; ced-5 55 _ -r - C' a. 35 - 0 30 - z 25 - -r 2015- 15- 10- 10- 5- 5- 0- B/C 1.5X 2X 3XE 3XL 0- Li B/C C 60 55- w 0. I ced-8 D 60 2X I 55- 50 - 50 - 45- 45- 40- 40 3XE 3XL Li ced-7; ced-5; ced-8 -r - C 35 - 35- 0 30- o 30 25- z6 25- z0 1.5X Developmental stage Developmental stage 20- 20- 15- 15- 10- 10- 5 50. 0 B/C 1.5X 2X 3E 3XL Developmental stage L1 T BIC 1.5X 2X 3XE 3XL Developmental stage Li Figure 2. Delayed onset of DNA degradation in ced-8 embryos. (A) N2 (B) nuc-1(e1392) (C) ced-8(n1891) (D) unc-10(e102) ced-8(n1891) nuc-1(e1392) egl15(n484). The y axis represents the number of TUNEL-positive cells present in embryos. Stages examined: bean and comma-stage embryos (B/C); 1.5-fold embryos (1.5X); and 3-fold embryos including both early and late stages (3X). Error bars indicate one standard error of the mean (sem). At least 24 embryos of each stage were examined. 78 Figure 2 A 5248- U) 0 0. z B wild type 5248- 44- 44- 40 - 40- 36 - 36- 32 - 32- 28 - 0 9. 24 - z 20 12- z 0 2420128- 84- 28- 16- 16 - 6 z 4- I - B/C - I 1.5X W 0- B/C 3X 52- D ced-8 48- 5248- 40 - 40- 36 - 36- 28 - 0 z 20- 2016- 16- 6 z 12- 128- 8- 4- 40 28 24- 24 - 6 z ced-8 nuc-1 32- 32- z 3X 44- 44- 0 1.5X Developmental stage Developmental stage C nuc-1 . - - - B/C 1.5X 3X Developmental stage 0- B/C 1.5X 3X Developmental stage Figure 3. ced-8 cloning and cDNA sequence. (A) Cosmids assayed for transformation rescue. Rescuing cosmids are in bold. (B) Subclones assayed for transformation rescue; the fraction of assayed transgenic lines showing rescue of the Ced-8 phenotype is indicated on the right. A, Agel; H, HpaI; M, MluI; N, NheI. (C) A composite of the C. elegans ced-8 sequence derived from cDNA and 5' RACE analysis. Intron positions are indicated by vertical bars. The predicted CED-8 protein sequence is shown beneath. Arrows designate base changes in the indicated ced-8 alleles. The ced-8(n3113) allele (*) is predicted to eliminate the splice acceptor for intron 3, resulting in an in-frame insertion of the sequence VTHFLGSVRKTKSDLQ into the CED-8 protein between amino acids 137 and 138. The ced-8(n2090) allele (t) contains two nucleotide changes relative to the wild-type sequence. Two in-frame stop codons upstream of the predicted initiation codon are underlined; the termination codon is also underlined. 80 Figure 3 B A Rescue? F54F4 C04B10 ClOAll C02B8 C51D5 F54F4 F37F1 F16F9 K11G11 C36A10 KO1FlO T01B10 F13B9 C46D5 7/7 A5/5 pGS19 N fpGS22 I pS22 nP81 40kb M pGS17 C03G5 C35H6 pGS23 1 3/3 ii___ N H i. 4kb 5/5 . C CAGACGCACATCATCCAACCCCAGTGGTCCTGAATATACACCACACATACTGTTCAGTTTTTATCGATTTGTTTAATTTAATATTTTTCGTCTAGTAAGCTACAATGTTTCTCAAAAAACACAAGTCAAAACTTTTGTTAGTTCC M F L K K H K S K L L L V P ATGAAGAACAAC;AAGATGCAGGAATTGTCGCAGTACTCACAGATAGAATCCCAAGTGTGCTTCTTGTCAGATGGTTTGACCTTTTCTGCTTTGGATTTGCAATGTGCTCCTATGCTCTTGATTTTTTCTCGGATATAGGTATTGC E E Q E D A G I V L T An3245 GE A V A D R I P S V L L V R W F D L P C F G F T n1999 SNL A A M C S Y A L D F F S D I G I A TTCATTTTTGGGCGGGAAGAIrACCTCTCCGGGTCACTGGTCCTGGCTTTTGCATTGCTACCTTCTGTGATTATTAACATAATTTCAATGGTTTGGATGCTCGATGACGAGATGCATTGGAAAAGAAGAGCTCATCCAAGAAGAAC H F W A G R Y L S G S L V L A F A L L P S V n31131 I I I N An3115 G1R AA I S M V W M L D D E M H W K R R A H P R R T An2090t Wb-stop CATTTGAGCTGAATCAGAAAAGGTTTATACCTTTAAGCAAAATGATCGTGCTGTGTATTTGCCAG TGGGACCACTCTTCTGGTACTACAAAGCTCTATACTACGGATGGATGTTCAGAAAAAGTAGTAACGAAAACACTGATGG F E L F I P L S K M I V L C I C Q M G P L F W Y Y K A L Y F Y G W M A n3313 GhE A R K S S N E N T D G R K S K M V C F Anlu9I Whutop E A E R D A T L L R F F E A F L E S A P Q L I I Q G A S Y F Q N Y Y Q T G N Q K R S I A A TCCTTATTGGTTGTATTTCCAAGCCGCATCTCTATTACTTTCAATCATTAGCATTTCCTGGTCCGTCGTTGTTCAAAACCGCTCGTTACGAATGATTAGGGATGACAAAGTCAATATTTGGCCACACGAAGCGGTTTTGCAGrT P Y W L Y An3244 Wtop F Q A A S L L L S I I S I S W S V V V Q N R S L R M I R D D K V N I W P H E A V L Q F kn2090t AT GGAGATTCCTCACGATTTTAGCGAGGATTATCACACTTGTCGCATTGGTGCTGATATTTGGTATCAATGTGGTACCTTTAATATCAGTTCATTTATTGGTTACGCTAGTTCACGTAATTTTCCTTCAzPCAATCCACATAGATGC R F L T I L A R I I T L V A L V L I F G I N V V P L I S V H L L V T L V H V I F L Q A I H I D A A n2093 EhK CTCATATTGAGAAGCTGCTCCTTCTTATCAATACTTTCATCCATATTTTTATACCGTTCAATATGGTGGAAGGAAACACACGGTGGCGATATTTAACAGCCTATTCTGTTGAGTTTATTGAGATGATG|TAGTCTGCTGGCTTCT H I E K L L L L I N T F I H I F I P F N M V E G N T R W R Y L T A Y S V E F I E M M L V C W L L TGTCATTAAATACATTTCCGTATATTGAAAAAGTTCAAGTTGGCGTTCCAATTTCATTTATAGCTG TATTGCAATCATGATGATGTATTATCAGTTTTTCCATCCCAACAGACGGCAGCTAATTGTTACGCAATCACAAGAAGA S L N T F P Y I E K V Q V G V P I S F I A G I A I M M M Y Y Q F F H P N GTCTGAATGTGCAGAAATCCGTTGAGACGCTCACCCCGAAACTAGAAAGTTCATTGGAAATTAGTGGAGAACAAAATACGAGTCAAGATTTGGTATCAGAATTACTTCTT S V E T L T P K L E S S L E I S G E Q N T S Q D L V S E L L L L N V Q K CTGTTGTCGGAAAACGAGTTCATTACTACATATTATCATAAAAATTGATATTAAATAAATAAATAAATGCAAAGTC R R Q L I V T Q S Q E D GATGTGGAACATGAAAATTAACAAACTAAATTAC D V E H E N Figure 4. ced-8 developmental northern blot. Top panel, the ced-8 transcript (1.4 kb) is expressed in embryos and to a lesser extent in LI larvae. YA, young adults. Bottom panel, the same blot probed with cyt-1 (Hengartner and Horvitz, 1994b), which is expressed at equal levels throughout development. 82 Figure 4 A .0 1.4-- cyt-1 Figure 5. CED-8 protein sequence comparisons. (A) Alignment of the full-length proteins C. elegans CED-8 and human XK. Identities are shaded black; similarities are shaded grey. Predicted transmembrane-spanning domains are indicated with brackets. Transmembrane-spanning domains in XK are as in Ho et al. (1994). (B) Kyte-Doolittle hydropathy plots (Kyte and Doolittle, 1982) (scanning window, 7 amino acids) of CED-8 and XK. (C) Alignment of similar sequences of CED-8, XK, Ciona intestinalisCOS41.5, and DrosophilaEST LD13628. 84 Figure 5 A KH 1 M F L K CED-8 S K K L L L V XK CED4 XK IRY FWA I G I A IFH 31 - - - - - - - -61 121 Q K 62 L S CEDXK SG V WQ TILEi E" CK 224'IA S I L L 170 K E V GQ A E 30 D E M H W K R R A H P R R T G T F E L N - - - - - - - - - - - - - - - - - - - - 120 E R L S S Q 61 - - - - - - - - 172 C F S S N E N T D G W Y 1,K A L Y Y G W M F R I T G N N E E P Y V CE V F C I YjF Q T H RSAF L T _ _ V 290 - - - - - - - - - - IEF C W S A V Q L K I D S ND L CED-8 XK 376 K-Q V 350 L G Q CED-8 428 PKLES XK 410 K P G L SI F MP N G L S E S fl QN' T P R D R D E I E 119 - F PEN IE F W C S G S P fL LLLD G Q F 3C - - - VY L M Q L I V R L F S S S K T S P H E ID L 281 K NflF fl p Q V N A w L w y P L S F K T Dl N TIP MV M N VM S V - - - - - - - - E D L P C G F Q R W L R C F C WIC R Q Y I E 375 C A P 349 P G 409 40L 458 E HfN N A P 444 D L C S A C 5.00 4.00 3.00 52.00 181 CED-8 137 XK 9 COS41 .5 LD13628 187 _iiz1 r1r1 1i 1.00 t 0.~.00 X3.00 sm wIllm 'V '1 -4.00 -6.00 5.00 53 240 11) 3O 4! D 0 3 - 4.00 -- 3.00 .- 2.00 .4 -L N1.00 ,.-Z2.00 v XK1.00-i 100 .. r sq if - - r vi -400 91 I&0 1d0 20 A0 0 30 4 0 289 289 Y*G L C FT I SR NVG Y F1229 L FLTEA - V E GUTR W WY S K S QDLVS TYW NY YY Q T jWI EALQC G -SI AI L F A C LEIS I H Y P WI F S F F I 329 D L Q G I O __V'IR CED-8 XK E SG RASVI A GSL CED-8 282 V L'F L K T W. 230 T S XK CE"= - E 173 B V W M L F V H R IS T L. R T ,S KS H LD60 GF F DL VRW V I V A A C - - D R CED-8 XK 120 D R G SK FI P R D E E Q E D A G I V A V L T D M 1 RA S M TI Y A D F S 383 CED-8 359 XK COS41 .5 271 G GS Yf F L C V S 202 158 30 208 A I 404 F 378 292 S Figure 6. ced-8:gfp expression (A) Embryo in which a ced-8:gfp fusion was expressed in most if not all cells of early embryos and is localized to the plasma membrane. (B) Nomarski image of the same embryo. 86 7=q Figure 6 Chapter 3 ced-li is required for the apoptotic morphology of programmed cell deaths in C. elegans Gillian M. Stanfield, Erika Hartwieg & H. Robert Horvitz Electron microscopy, from fixation to photography, was done by Erika Hartwieg. This chapter is in preparation for submission for publication. 88 Summary We identified ced-11 in a screen for mutations that alter the appearance of programmed cell deaths in C. elegans. In ced-11 mutant embryos, the morphology of programmed cell deaths is abnormal as visualized by Nomarski microscopy and electron microscopy. These abnormal-looking dying cells appear to be engulfed by normal mechanisms. ced-11 acts downstream of the cell-death execution gene ced-3 to effect aspects of the conserved apoptotic morphology of programmed cell deaths. We cloned the ced-11 gene and found that it encodes a protein similar to members of the TRP family of calcium channels, implicating calcium signaling in the downstream events of programmed cell death. 89 Introduction Programmed cell death is a conserved feature of metazoan development and homeostasis (reviewed by Jacobson et al., 1997; Vaux and Korsmeyer, 1999). As animals and plants grow, many cells are generated only to be eliminated by programmed cell death as development continues. During development, programmed cell death is important for morphogenesis and for creating matched populations of cells, as in the mammalian nervous system (reviewed by Jacobson et al., 1997; Pettmann and Henderson, 1998). In adults, programmed cell death is important for maintaining tissue homeostasis, and failure to regulate cell killing results in disease (reviewed by Thompson, 1995). The process by which cells die, both in diverse species and in response to diverse cues within a single species, appears morphologically similar and has been termed apoptosis (as distinguished from the death of cells caused by physiological damage, called necrosis) (Kerr et al., 1972; Wyllie et al., 1980). A cell dying by apoptosis undergoes a series of morphological changes that affect nearly all aspects of its structure, as visualized by light or electron microscopy (EM). The plasma membrane exhibits blebbing or ruffling, the cytoplasm shrinks, the nuclear envelope dilates and breaks down and chromatin condenses at the periphery of the nucleus. Relative to most living cell types, dying cells label heavily with electron-dense stains when analyzed by EM. Organelles remain intact at these stages of cell death. The cell corpse eventually fragments into smaller membrane-bound bodies, which are engulfed and then degraded inside the engulfing cell. Importantly, apoptotic cell deaths do not lead to damage of surrounding tissues, since the cell corpse remains intact or enclosed as membrane-bound fragments and is rapidly cleared from live tissue by phagocytic activity. Other common features of apoptotic cells can be assayed by specific staining and labeling techniques. Certain vital dyes (e.g., acridine orange) are excluded from viable cells but accumulate within dying cells (Saunders et al., 1962; Abrams et al., 1993). Phosphatidylserine is normally confined to the inner leaflet of the plasma membrane but becomes exposed in the outer leaflet when cells die (Fadok et al., 1992); this exposure can be detected via binding of fluorescein-coupled Annexin V (Martin et al., 1995). The chromosomal DNA of dying cells is degraded by endonuclease activity; cellcorpse DNA appears as an ladder of internucleosomally cleaved fragments when 90 analyzed by gel electrophoresis or can be labeled in situ using the TUNEL (terminal transferase-mediated dUTP nick end labeling) technique, which labels cell corpses by virtue of their increased number of DNA ends (Gavrieli et al., 1992). Since the morphologies of diverse cell types undergoing apoptosis are similar, it is plausible that the underlying mechanisms of killing are also conserved. Much attention has been focused on the genetic and biochemical control of programmed cell death, and a central pathway for cell killing has been established that indeed is conserved in organisms as diverse as mammals and the nematode Caenorhabditiselegans. In C. elegans, the activities of three genes, egl-1, ced-4 and ced-3 (egl, ejgg-aying defective; ced, cell death abnormal), are required for cell killing (reviewed by Metzstein et al., 1998). In living cells, the cell-killing activities of ced-4 and ced-3 are inhibited by the function of the gene ced-9 and in dying cells, egl-1 inhibits ced-9 function, allowing the ced-4-mediated activation of ced-3. ced-9 encodes a member of the bcl-2 family of celldeath regulators, and egl-1 encodes a BH3 (bcl-2 homology region 3) domain-only member of the bcl-2 family. ced-4 encodes a protein similar to mammalian Apaf-1 (apoptosis-activating factor-1). ced-3 encodes a caspase (cysteine aspartate-specific protease) and is likely to be the primary effector of programmed cell death in C. elegans. In most cell types examined, caspase activation and (presumably) cleavage of target proteins is necessary for programmed cell death and its concomitant morphological and structural changes (reviewed by Thornberry and Lazebnik, 1998). While much progress has been made in understanding the molecular mechanisms by which cell death is initiated, the link between these key cell-death regulators and most aspects of the morphological changes that cells undergo as they die is not clear. Defects in specific aspects of the morphological changes of dying cells linked to an absence of caspase-3 have been observed in dying mammalian cells (Porter and Janicke, 1999). In a caspase-3 defective cell line (MCF-7), programmed cell deaths can be induced but membrane blebbing, cell shrinkage and DNA degradation do not occur. Transfection of caspase-3 into this cell line rescues these defects (Janicke et al., 1998). While many caspase cleavage targets have been identified (reviewed by Stroh and Schulze-Osthoff, 1998), their roles in the death process are mostly unexplored. In mammalian cells, mitochondria have been implicated in the killing step of programmed cell death (reviewed by Kroemer et al., 1998). When cells die, there is a reduction in the mitochondrial transmembrane potential associated with opening of the 91 permeability transition pore (PTP), loss of mitochondrial homeostasis and the release of pro-apoptotic factors, AIF (apoptosis-inducing factor) and cytochrome c, which promote caspase activation and possibly additional caspase-independent killing functions. Some aspects of cell-death morphology can be induced in the presence of caspase inhibitors (Deas et al., 1998) and therefore may be induced by mitochondria, but the link between these mitochondrial events and downstream morphological changes of cell death is not clear. We have performed a genetic screen to isolate C. elegans mutants defective for aspects of the programmed cell death process and identified the gene ced-11, the function of which is necessary for the conserved morphology of programmed cell deaths but not required for cell killing per se. CED-11 may function as a downstream component of the cell-death pathway. We have cloned the ced-1 1 gene and found that it encodes a protein with similarity to members of a family of ion channels typified by the DrosophilaTRP (.ransient receptor potential) calcium channel, implicating calcium signaling in the conserved series of morphological changes that occur in dying cells. 92 Results A screen for cell-death mutants During the development of the C. elegans hermaphrodite, 131 of the 1090 somatic cells generated die, 113 of these during embryonic development (Sulston and Horvitz, 1977; Sulston et al., 1983). Most cell divisions occur during the first half of embryogenesis, and most programmed cell deaths occur soon after cells fated to die are generated. As visualized by Nomarski differential interference contrast microscopy, a dying cell acquires a raised, refractile appearance. After a cell corpse is engulfed, usually within an hour of becoming refractile, it is no longer visible by light microscopy (Sulston and Horvitz, 1977). In animals mutant for any of the engulfment genes ced-1, -2, -5, -6, -7 or -10, many persistent refractile cell corpses remain visible for hours or even days after these cells have died (Hedgecock et al., 1983; Ellis et al., 1991). We used the presence of persistent corpses in an engulfment-defective mutant as the basis of a genetic screen for animals defective in programmed cell death during embryogenesis (Fig. 1). We performed our screen similarly to that of Ellis et al. (1991) with two modifications. First, we sought animals containing either more or fewer cell corpses than are present in the engulfment mutant ced-5(n2098), which has an intermediate engulfment-defective phenotype relative to other known engulfment mutations (Ellis et al., 1991 and G.M.S. and H.R.H., unpublished observations; Wu and Horvitz, 1998), rather than looking for the presence of corpses at later stages than in the wild type. Second, we examined embryos, whereas previously only larval stages had been examined. Like Ellis et al. (1991), we performed our screen using a sem-4 mutant background and Nomarski microscopy to examine collections of F3 progeny of mutagenized hermaphrodites (to be able to isolate maternal as well as zygotic mutations). sem-4 mutant animals lack sex muscles and thus are egg-laying defective (Basson and Horvitz, 1996), and their progeny develop and eventually hatch inside the mother's body. This approach allowed us to examine collections of embryos from the same mother, possibly increasing the chance of identifying weak or incompletely penetrant mutations and allowing recessive mutations causing lethality to be isolated from heterozygous siblings. We screened approximately 15,000 mutagenized haploid genomes for zygoticallyacting genes and 6,000 haploid genomes for maternal-effect genes. We isolated 12 93 mutations with specific recessive effects on programmed cell death. Seven mutants had few or no cell corpses in embryos of any stage, consistent with a block in programmed cell death. We found that these mutants represented two alleles of ced-4, n2723 and n2831, and five alleles of ced-3, n2719, n2720, n2721, n2722 and n2830 (data not shown; S. Shaham, B. Davies, P. Reddien and H.R.H., unpublished results). The other five mutants had a similar novel cell-death phenotype, described below. They defined one complementation group, the cell-death gene ced-1 1. Cell corpses appear abnormally non-refractile in ced-11 mutant embryos As visualized by Nomarski microscopy, the five ced-11; ced-5 strains isolated in the screen all contained a reduced number of refractile cell corpses in embryos as compared to the ced-5 strain (Fig. 2A). Furthermore, these strains contained non-refractile structures that were not present in wild-type C. elegans embryos (Figs 2B, 3). These nonrefractile structures varied in appearance from an abnormally rounded nucleus, to a detached-appearing rounded nucleus, to a hole in the tissue with debris floating inside (Fig. 3). Four of the five mutant strains isolated in the screen contained few if any refractile cell corpses in embryos, and these non-refractile structures became more refractile at about the time of hatching (data not shown). The fifth strain, ced-11(n2834); ced-5, contained both refractile cell corpses and the non-refractile structures (Fig. 2), suggesting that ced-11(n2834) is a weak ced-11 allele. Larvae of all five strains contained both refractile cell corpses and the non-refractile structures. The phenotype caused by ced-11 mutations is recessive and is not maternally rescued: approximately one-quarter of the progeny of ced-11(n2744)/+; ced-5 animals exhibited the Ced-11 phenotype (data not shown). Animals trans-heterozygous for ced-1 1 (n2 744) and the deficiency qDf2 looked similar to ced-1 1 (n2 744) homozygotes (data not shown), suggesting that ced-11(n2744) causes loss of ced-11 function. ced-11 mutant strains are viable and fertile and exhibited no obvious non-cell death-related defects (data not shown). The pattern of the abnormal non-refractile structures in ced-11 embryos was similar to that of refractile cell corpses in wild-type embryos: the structures became visible at the time during embryogenesis when most cell deaths occur, and most were present in the head region, where most cell deaths occur in wild-type animals and cell corpses are visible in engulfment mutants such as ced-5. Since the abnormal non-refractile 94 structures apparently replaced refractile cell corpses in the embryo and could become refractile, or corpse-like, at about the time of hatching, it seemed likely that the nonrefractile structures in ced-11; ced-5 animals were cells that normally undergo programmed cell death but in ced-11 instead appeared abnormally non-refractile. We determined whether mutations in ced-1 1 result in the survival of cells that normally die. To assay cell survival, we counted the number of cells in the anterior pharynx (Hengartner et al., 1992), a region where 16 cells die during wild-type development. We found that in ced-1 1 animals, there is no significant survival of cells that normally die in this region (Table 1). We also tested whether the ced-11(n2744) mutation might result in extra cell survival in a sensitized genetic background. For example, mutations in the gene ced-8 result in very little survival of cells that normally die but do enhance weak mutations in either ced-3 or ced-4 (G.M.S. and H.R.H., manuscript in preparation; see Chapter 2). In contrast to ced-8 mutations, ced-11(n2744) did not enhance the cell-survival phenotype of the weak ced-3 allele n2427 (Table 1). Furthermore, ced-11(n2744) did not enhance the very weak cell-survival phenotype of ced-8(n1891) (Table 1). To determine whether the nonrefractile structures were cells that normally undergo programmed cell death, we determined whether strong ced-3 or ced-4 mutations, which block nearly all programmed cell deaths, would eliminate the abnormal non-refractile structures in ced-1 1 mutants. Since very few cell corpses are visible in ced-1 1 single mutants (described below), we used a ced-5 mutant background to allow the visualization of as many cell corpses as possible. We found that almost no cell corpses or abnormal non-refractile structures were visible in ced-11; ced-3 ced-5 mutant animals (Table 2). We also found that no refractile cell corpses or non-refractile structures were visible in ced-4 ced-11 embryos (Table 2). Thus, the pattern of the non-refractile structures within ced-11 embryos was similar to the pattern of refractile cell corpses in wild-type embryos, and the presence of the non-refractile structures was dependent on the initiation of programmed cell death. These data suggest that the abnormal non-refractile structures in ced-1 1 mutants are cell corpses and that ced-1 1 activity is required for the normal refractile appearance of the corpses of cells dying by programmed cell death. Although these data establish that cell deaths in ced-1 1 mutants are dependent on the activity of ced-3 and ced-4, they do not distinguish whether all aspects of cell killing are normal. In particular, it is not clear 95 whether, like the refractile structures in wild-type animals, the non-refractile structures represent cells that are fully committed to die. Since we believe that the non-refractile structures likely represent "dead" cells, we will refer to them as "non-refractile cell corpses" throughout the text. Engulfment of ced-11 non-refractile cell corpses To determine whether the appearance of the non-refractile ced-11 cell corpses was dependent on the ced-5 background in which they were isolated, we examined the pattern of non-refractile and refractile cell corpses in animals mutant for ced-11 and the engulfment genes ced-1, -2, -6 or -10. We counted the number of non-refractile cell corpses present in the head of ced-1 1 engulfment ced double mutants at the 3-fold stage of embryogenesis, a time in development at which very few cell corpses are visible in wild-type animals but many persistent cell corpses are present in engulfment mutants. We found that many non-refractile cell corpses are present in 3-fold embryos mutant for ced-1i and any of these engulfment genes (Fig. 4). Mutations such as ced-2(el 752), with a weak effect on the engulfment of refractile cell corpses (Ellis et al., 1991), resulted in only a few persistent non-refractile cell corpses, and mutations such as ced-1 (el 735), with a stronger effect on the engulfment of refractile cell corpses (Ellis et al., 1991), resulted in many persistent non-refractile cell corpses. That the number of abnormal non-refractile cell corpses in ced-1 1 mutant embryos is increased proportionally to defects in engulfment suggests that these structures can be recognized and engulfed by the same machinery that recognizes and engulfs wild-type cell corpses. Very few non-refractile cell corpses were visible in ced-11 single mutants, and most were present during mid-embryogenesis, which is approximately when most refractile cell corpses are visible in wild-type embryos. However, the maximum number of nonrefractile corpses was visible slightly later than in wild-type embryos (Fig. 5A, B). We observed fewer non-refractile cell corpses in ced-11 mutants than there are refractile cell corpses in wild-type embryos. These corpses resembled abnormally round detached nuclei as opposed to holes with debris. Similarly, corpses present at early embryonic stages in ced-1 1; engulfment ced double mutant strains usually resembled abnormally round nuclei and more distinct structures and holes with debris were usually present at late embryonic stages. Thus, when non-refractile cell corpses are engulfed quickly, as in a ced-11 single mutant, they are difficult to distinguish from living cells, but when they 96 persist unengulfed, their appearance becomes more distinct from both living and normal dying cells. The delayed onset of cell corpses that we observed in the ced-1 1 single mutant could be caused by a delay in cell killing, a short delay in the engulfment of cell corpses (as opposed to the long delay or block in engulfment that results from mutations in the engulfment genes and causes the accumulation of cell corpses throughout embryogenesis, rather than for a short time period), or a bias in our observations toward detecting ced-1 1 cell corpses more frequently in older embryos (since the nonrefractile cell corpses appear more distinct at these later stages). To attempt to distinguish among these possibilities, we examined the accumulation of non-refractile cell corpses during embryogenesis in ced-1i; ced-5 animals. The appearance of nonrefractile cell corpses was slightly delayed relative to the appearance of refractile cell corpses in ced-5 mutant embryos, and slightly fewer of the non-refractile cell corpses were visible relative to the maximum number of refractile cell corpses in ced-5 embryos (Fig. 5C, D). Since we observed a delay in the onset of cell corpses even if engulfment was blocked, it is unlikely that the altered kinetics of the non-refractile cell corpses in ced-1 1 embryos is caused by a delay in engulfment. We cannot distinguish between a delay in the appearance of cell corpses and a bias in our detection of cell corpses. To summarize, our observations indicate that mutations in ced-11, which affect the morphology of cell corpses, do not affect cell-corpse engulfment. ced-11 mutations may have a weak effect on the kinetics of cell killing, although if so, this delay does not result in cell survival. Non-refractile ced-11 cell corpses are not electron-dense by EM To determine the ultrastructural basis for the abnormal appearance of cell corpses in ced-1 1 mutants, we used transmission electron microscopy to examine cell corpses in ced-1 1 animals. We chose to examine the corpses of cells that die in the posterior ventral cord during the L2 larval stage for four reasons. First, seven cells die in this region during a relatively short developmental time period (Sulston and Horvitz, 1977). Second, nuclei of the ventral cord form an approximately linear array along the ventral body wall of the animal (Sulston, 1976), facilitating the unambiguous identification of nuclei in serial sections. Third, a time-course study of the morphological changes in dying cells in this region has previously been performed (Robertson and Thomson, 97 1982), so the deaths of these cells in wild-type animals are particularly wellcharacterized. Fourth, both refractile and non-refractile cell corpses can be examined in ced-il larvae (see above). So that multiple cell corpses from a single animal could be examined, we used a ced-1 mutation to prevent the rapid engulfment of dying cells. ced-1 mutations delay cell-corpse engulfment but do not affect the morphology of cell corpses, which appear normal in ced-1 mutants both by Nomarski microscopy and by EM (Hedgecock et al., 1983). We examined cell corpses from wild-type, ced-1(e1735) and ced-10(e735); ced-11(n2744) larvae (see Materials and Methods). We found that, as previously reported (Robertson and Thomson, 1982; Hedgecock et al., 1983), wild-type and ced-1 cell corpses were indistinguishable from one another by electron microscopy and shared many aspects of the apoptotic morphology observed in diverse cell types from many organisms (Fig. 6A-D and data not shown). The cell corpses appeared small and rounded, consistent with shrinkage and the loss of normal cell-cell contacts. Condensed chromatin was visible within the nucleus, mitochondrial structure was not obviously different from that in living cells and both the nuclei and cytoplasm of cell corpses appeared darker (more heavily-labeled with electron-dense EM stains) than those of surrounding cells. One common feature of mammalian cell corpses, the fragmentation into smaller membrane-bound bodies (apoptotic bodies) has not been seen in C. elegans (Robertson and Thomson, 1982) and we did not observe any such fragmentation. Most aspects of apoptotic cell-corpse morphology appeared to be conserved in nonrefractile ced-iI cell corpses (Fig. 6E, F and data not shown). These cells were small and rounded, and neither chromatin condensation nor mitochondrial morphology were obviously different from what occurs in wild-type cell corpses. However, unlike wildtype cell corpses, ced-1 1 non-refractile cell corpses were not darker than surrounding cells and were stained less densely than were normal cell corpses. Thus, the absence of refractility in ced-1 1 cell corpses appeared to correlate with the absence of dark, electron-dense staining in EM sections. ced-11 cloning We cloned the ced-1 1 gene using a positional cloning approach. We genetically mapped ced-1 1 on LG III to the region between unc-32 and ced-7 and very close to glp-1 (Fig. 7A; see Materials and Methods). We injected DNA clones from this region into ced-1(el735); 98 ced-11(n2 744) mutant hermaphrodites and scored embryos for refractile cell corpses, the presence of which would indicate rescue of the Ced-11 mutant phenotype. We found that an approximately 12 kb subclone of cosmid C33D7 containing a single predicted gene, ZK512.3, rescued the Ced-11 phenotype (Fig. 7B, C). By isolating ZK512.3 cDNAs and performing 5'-RACE, we confirmed the structure of the gene as predicted by GENEFINDER (L. Hillier and P. Green, personal communication) (Fig. 7D) and determined that ZK512.3 is SL1 trans-spliced(Krause and Hirsh, 1987). The ZK512.3 cDNA is predicted to encode a protein of 1418 amino acids with at least seven hydrophobic, possible transmembrane-spanning regions (see Fig. 9A, C). To test whether ZK512.3 corresponds to ced-1i, we placed a full-length cDNA under the control of C. elegans heat-shock promoters and found that expression of ZK512.3 rescued the Ced-11 mutant phenotype (data not shown). We determined the sequence of the coding region and intron-exon junctions of the five ced-1 1 alleles and found lesions in all five (Fig. 7D), further confirming that ced-1 1 corresponds to ZK512.3. We also determined the sequence of a sixth allele, w23 (isolated as a spontaneous mutant and kindly provided by J. Rothman), which caused a phenotype similar to that caused by the four strong ced-11 alleles isolated in our screen and failed to complement ced-1 1 (n2744). The ced-1 1 (w23) allele contains a five bp deletion and should generate a product consisting of the first 77 amino acids of CED-11 followed by three out-of-frame amino acids. To determine when ced-11 is expressed during development, we probed a northern blot of RNA samples from developmentally-staged wild-type animals with the ced-1 1 cDNA. We observed a single band of approximately 4.2-4.7 kb in embryos and Li larvae, which was consistent with the size of the 4.4 kb transcript predicted by cDNA sequencing and 5'-RACE analysis (Fig. 8). CED-11 is weakly similar to TRP proteins CED-11 shares similarity with two mammalian (TRPC7 and melastatin) and three C. elegans (TO1H8.5, F54D1.5 and C05C12.3) proteins, which form a distinct subfamily within the TRP family of ion channels. The TRP proteins are typified by Drosophila trp (transient receptor potential), which is involved in visual transduction. At least some TRP-family members are thought to function in the process of store-operated calcium entry (Putney and McKay, 1999). A sequence alignment of CED-11 and the two human proteins is shown in Fig. 9A. 99 The defined features of TRP proteins are six presumed membrane-spanning domains and a pore region and a set of amino-terminal ankyrin repeats (Putney and McKay, 1999). The CED-11-like subfamily is most similar in sequence to other TRP channels in the putative transmembrane and pore-forming region. The highest similarity between CED-11 and other members of the CED-11-like subfamily also lies in this region, although there is similarity throughout the protein sequences (Fig. 9B). CED-11 shares approximately 20% identity and 40% similarity with the human proteins in the most highly conserved region (Fig 9C). The three C. elegans predicted proteins are more similar to TRPC7 and melastatin than is CED-11 (Nagamine et al., 1998), so CED-11 is the most divergent member of this group. 100 Discussion ced-11 controls cell-corpse morphology C. elegans programmed cell deaths are morphologically similar to mammalian apoptotic cell deaths. Mutations in the ced-11 gene cause dying cells to appear non-refractile as visualized by Nomarski optics and non-electron-dense as visualized by EM. The basis for the dark staining of cells dying by programmed cell relative to living cells is not well-understood, but may be due partially to intracellular acidification, which should increase the affinity of the tissue for the uranyl acetate and lead citrate stains used in EM procedures (Hayat, 1970). Ultrastructurally, ced-1I non-refractile cell corpses did not display the vacuolation that characterizes damaged cells dying by necrosis in mammals (Wyllie et al., 1980) or the membranous structures seen in C. elegans neurons expressing cytotoxic forms of ion channels (Hall et al., 1997). These observations are consistent with the idea that ced-1I mutations do not disrupt cellular homeostasis, as do these non-apoptotic forms of cell death. Rather, ced-11 mutations seem to affect specific aspects of apoptotic morphology. The non-refractile cell corpses of ced-11 mutants were slightly delayed in appearance and reduced in number compared with the refractile cell corpses seen in wild-type animals. This effect of ced-1 1 mutations is weaker than that of ced-8 mutations, which result in a more pronounced delay in cell-corpse appearance (G.M.S. and H.R.H., manuscript in preparation; see Chapter 2). Furthermore, ced-8 mutations weakly inhibit cell killing and enhance weak alleles of ced-3 or ced-4 to result in the survival of many extra cells. That mutations in ced-11 apparently did not result in the survival of cells that normally die, even in sensitized genetic backgrounds, suggests that the altered pattern might not result from a defect in the killing process per se. Nonrefractile cell corpses are very difficult to see initially and become clearly distinct from living nuclei only if they persist in the embryo (e.g., in engulfment-defective animals). It is possible that some ced-11 cell corpses are engulfed without first becoming distinguishable from living nuclei by Nomarski microscopy. To confirm that cells die with normal kinetics in ced-1 1 animals, it may be useful to examine the pattern of appearance of cell-corpse markers other than refractility in ced-1 1 embryos. 101 ced-11 acts downstream in the genetic pathway for programmed cell death ced-11 non-refractile cell corpses were engulfed normally and were under the control of the known engulfment genes, indicating that ced-1 1 mutations specifically alter cellcorpse morphology. Mutations in ced-3 block cell deaths in ced-11 animals, indicating that ced-1 1 functions downstream of ced-3 killing. That cell-corpse engulfment and cellcorpse morphology are genetically separable suggests a model in which different downstream events in cell death are activated independently (Fig. 10). The CED-3 caspase presumably initiates these downstream events via cleavage of specific substrates. Some of these substrates may be activated by cleavage, while others may be inactivated. Since loss of ced-1I function results in morphological abnormalities, if CED-11 is regulated by the CED-3 caspase in C. elegans, it must be activated during programmed cell death. This activation could occur either directly, by cleavage of the CED-11 protein, or indirectly, e.g., by association with a CED-3-activated protein. In the presence of mutations that block the engulfment process, persistent nonrefractile cell corpses become refractile at about the time of hatching, suggesting that ced-1 1 functions in embryos but not at later developmental stages. The transience of the nonrefractile cell corpses might result from genetic redundancy: another gene also could act to cause the refractility of cell corpses so that cell corpses still can become refractile in ced-11 embryos. What biological role might ced-11 serve? One clue is offered by our observation that in ced-11; engulfment ced double mutant animals, holes containing debris, likely derived from fragmented cell corpses, are sometimes visible. It is possible that ced-1 1 cell corpses are more fragile than normal cell corpses and thus more prone to breakage. It has been suggested that the release of the contents of a dying cell into living tissues could be deleterious (Ren and Savill, 1998). If so, ced-1 I might act to stabilize cell corpses and thereby prevent damage to surrounding tissues. Whatever the biological function of ced-11, the discovery of the gene offers a glimpse into the events of cell death downstream of the global regulators of cell killing in C. elegans. So far, the cell-death pathway has turned out to be remarkably conserved between nematodes and humans, and we suggest that ced-11, like the upstream cell death-regulatory genes, is conserved as well. 102 CED-11 might function as a calcium channel The CED-11 protein shares similarity with a subset of the TRP family of proteins, which are candidates to mediate store-operated, or capacitative, calcium currents (reviewed by Scott and Zuker, 1998; Putney and McKay, 1999). In this process, depletion of internal calcium stores results in the opening of calcium channels in the plasma membrane, allowing the generation of long-duration calcium signals. The similarity among CED-11, TRPC7 (Nagamine et al., 1998), melastatin (Duncan et al., 1998) and the C. elegans predicted proteins extends throughout the length of CED-11, most strongly in the predicted transmembrane and pore-forming region. However, CED-11 is clearly the most divergent member of this group of proteins. If CED-11 does function as a channel, its properties could well differ from those of the other proteins. Furthermore, none of the CED-11 subfamily of TRP channels has yet been shown to function as a calcium channel, so functional analysis is necessary to determine if the similarity between CED11-like proteins and TRP channels is meaningful. Nonetheless, this similarity indicates that it could be worthwhile to explore a role for calcium signaling during the downstream events of programmed cell death. Calcium is very important in diverse cellular processes and has long been implicated in diverse aspects of both apoptotic and non-apoptotic cell death (reviewed in Berridge et al., 1998; Lipton and Nicotera, 1998; Nicotera and Orrenius, 1998). Longterm elevations of calcium levels can result in cytotoxic (non-apoptotic) cell death, and treatment with calcium ionophores can result in apoptotic cell death. Additionally, increases in calcium concentration within the dying cell have been observed in apoptotic deaths triggered by various stimuli (Kaiser and Edelman, 1977). During the apoptotic deaths of mammalian cells, there is a reduction in the mitochondrial membrane potential and a loss of mitochondrial homeostasis associated with the opening of a large channel, the permeability transition pore (PTP), in the mitochondrial membrane (Kroemer et al., 1998). Increases in intracellular calcium levels can induce opening of the PTP, and PTP opening releases Ca" stored within the mitochondria. Thus, within a dying cell, increased calcium levels, whenever they occur in the death process, might function as a positive feedback mechanism to enforce commitment to cell killing via mitochondria. A number of the enzymes that are postulated to be involved in downstream celldeath events are calcium-dependent (reviewed by McConkey, 1996). These enzymes 103 include transglutaminase, phospholipid scramblase and some candidate cell-death endonucleases. Thus, the increased Ca" level thought to exist in dying cells could promote multiple aspects of cell-corpse formation, engulfment and degradation. Mutations in ced-1 1 primarily affect cell-corpse morphology rather than cell killing or cell-corpse engulfment and might reduce but not eliminate a calcium signal, thus affecting only those processes most sensitive to reductions in calcium levels. We favor a model in which ced-11-mediated calcium signaling is important only for events of cell death downstream of and triggered by CED-3 caspase activity. 104 Materials and Methods Nematodes Nematodes were cultured as described by Brenner (1974). All strains were derived from the wild-type Bristol strain N2. sem-4(n1378); ced-5(n2098) animals were mutagenized in 50 mM EMS as described by Wood et al. (1988). Worms used for RNA preparations were grown in liquid culture essentially as by Wood et al. (1988). All mutations are described by Riddle et al. (1997), except ced-5(n2098) (Wu and Horvitz, 1998). A list of mutations used follows. LG I: sem-4(e1378); ced-1(el735) LG III: lon-i(e185); dpy-17(e164); dpy-18(e364); dpy-19(e1259); unc-36(e251); unc-32(e189); unc-50(e306); unc-69(e587); glp-1(q231); ced-7(nl892); ced-6(n1813); ced-4(nl162) LG IV: dpy-19(e1282); dpy-20(e1282); ced-2(e1752); ced-3(n717, n2427); ced-5(n2098, n1812); ced-10(n1996) LG X: ced-8(nl891); nuc-1(e1392) Cell death assays Live animals were staged developmentally by appearance according to Wood et al. (1988). For corpse counts, the number of refractile cell corpses and/or non-refractile structures in the head of living 3-fold stage embryos was counted using Nomarski (differential interference contrast) microscopy (Ellis et al., 1991). For cell-survival counts, the number of cell corpses in the anterior pharynx of L3 and L4 larvae was counted as by Hengartner et al. (1992). TUNEL assays were performed as described by Wu et al. (manuscript in preparation; see Chapter 4). Genetic mapping ced-11(n2744) was placed on LG III by linkage to unc-32: 0/17 Ced-11 progeny of unc-32/ced-11; ced-5 mothers segregated Unc progeny. All subsequent mapping of ced-11 was performed using the n2744 allele. Other alleles isolated in the screen were tested for failure to complement n2744 and for linkage to unc-32 III. We found that w23 was allelic to ced-11 by genetic complementation and confirmed this result by allele sequencing. 105 ced-11 was mapped to the glp-1 region by the following data. From dpy-17 unc-69/ced-11; ced-5 heterozygotes, 11/17 Dpy non-Unc and 9/16 non-Dpy Unc progeny segregated Ced-11 embryos. From unc-69 dpy-18/ced-11; ced-5 heterozygotes, 13/13 non-Unc Dpy and 2/13 Unc non-Dpy progeny segregated Ced-11 embryos. From dpy-17 unc-36/ced-11; ced-5 heterozygotes, 21/21 Dpy non-Unc and 0/27 non-Dpy Unc progeny segregated Ced-11 embryos. From dpy-19 unc-32/ced-11 heterozygotes, 0/8 non-Dpy Unc and 10/10 Dpy non-Unc progeny segregated Ced-11 embryos. From dpy-19 unc-50/ced-11 heterozygotes, 16/22 non-Dpy Unc progeny segregated Ced-11 embryos. Finally, the following two crosses confirmed the proximity of ced-1 1 to glp-1 but did not place it either to the left or right side. First, of 67 non-Glp Unc progeny of glp-1 ced-7 unc-69/ ced-1I heterozygotes, 44/67 segregated Ced-11 Ced-7 embryos and 23/67 segregated Ced-11 embryos. Second, 22/22 Unc-32 non-Glp progeny of unc-32 glp-1/ ced-1 1 heterozygotes segregated Ced-11 embryos. Rescue experiments Injections were performed as by Mello et al. (1991). We coinjected DNA to be assayed for rescue at approximately 20 ng/gl with the plasmid pRF4 at 50-75 ng/gl into animals of the genotype ced-1 (el 735); ced-11(n2744) and established transgenic lines using the dominant Roller (Rol) phenotype conferred by the coinjection marker. We examined embryos using Nomarski microscopy for the presence of refractile cell corpses in the head (i.e., return to the Ced-1 phenotype). Since the Rol phenotype can not be scored before the L2 stage, we rescued embryos from slides and scored for the Rol marker 2 days later to confirm that transgenic animals had been examined. Electron microscopy We examined L2 larvae using Nomarski microscopy and chose animals containing visible cell corpses in the posterior ventral cord for EM study. We photographed and sketched the positions of living and dead cell nuclei in the posterior ventral cord region of selected larvae. We rescued the larvae from slides and transferred them immediately to a drop of cold water in the center of a small Petri dish; this liquid was replaced promptly by a solution of fixative (0.67% OsO 4,0.67% glutaraldehyde, 0.1M sodium cacodylate pH 7.2), and the larvae were placed at 4'C in the dark to fix for 60-75 106 minutes. The larvae were washed three times in 0.1M sodium cacodylate. Some larvae were cut in half using a fresh scalpel blade and the tail was allowed to continue fixing in fresh fixative solution for an additional 2-3 hours in the dark. Samples were then transferred to a solution of 2% OsO4 and placed at 4'C overnight. The osmium solution was washed off and the worms were embedded in an agar block. The block was dehydrated in a graded ethanol series, resin-infiltrated in a mixture of epon and araldite and allowed to polymerize overnight at 60'C. Serial sections (50 nm) were cut with an Ultracut E and contrast stained with 1% uranyl acetate and lead citrate. The sections were photographed on a JEOL 1200CX at 80 kV. Molecular biology Standard molecular biology procedures were followed (Sambrook et al., 1989). The sequences of primers used for PCR amplification, sequencing of ced-1 1 mutants and 5'RACE are available upon request. DNA sequencing was performed on an ABI 373A sequencer. We isolated RNA from developmentally synchronized nematodes by standard methods and performed poly(A) selection using Fast Track (Invitrogen.) 5'RACE reactions were performed on embryonic RNA using the 5' RACE system (Gibco.) 107 Acknowledgments We thank B. James for DNA sequencing; A. Coulson and the C. elegans Genome Sequencing Consortium for DNA clones; B. Barstead and P. Okkema for C. elegans cDNA libraries; and J. Rothman for the ced-11(w23) allele. We also thank M. Alkema, M. Metzstein and P. Reddien for helpful comments on the manuscript. G.M.S. was supported by a Howard Hughes Medical Institute Predoctoral Fellowship. H.R.H. is an Investigator of the Howard Hughes Medical Institute. 108 References Abrams, J. M., White, K., Fessler, L. I., and Steller, H. (1993). Programmed cell death during Drosophilaembryogenesis. Development 117, 29-43. Basson, M., and Horvitz, H. R. (1996). The Caenorhabditiselegans gene sem-4 controls neuronal and mesodermal cell development and encodes a zinc-finger protein. Genes Dev. 10, 1953-1965. Berridge, M. J., Bootman, M. D., and Lipp, P. (1998). Calcium--a life and death signal. Nature 395, 645-648. Brenner, S. (1974). The genetics of Caenorhabditiselegans. Genetics 77, 71-94. Deas, 0., Dumont, C., MacFarlane, M., Rouleau, M., Hebib, C., Harper, F., Hirsch, F., Charpentier, B., Cohen, G. M., and Senik, A. (1998). Caspase-independent cell death induced by anti-CD2 or staurosporine in activated human peripheral T lymphocytes. Immunol. 161, 3375-3383. J. Duncan, L. M., Deeds, J., Hunter, J., Shao, J., Holmgren, L. M., Woolf, E. A., Tepper, R. I., and Shyjan, A. W. (1998). Down-regulation of the novel gene melastatin correlates with potential for melanoma metastasis. Cancer Res. 58, 1515-1520. Ellis, R. E., Jacobson, D. M., and Horvitz, H. R. (1991). Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditiselegans. Genetics 129, 79-94. Fadok, V. A., Voelker, D. R., Campbell, P. A., Cohen, J. J., Bratton, D. L., and Henson, P. M. (1992). Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207-2216. Gavrieli, Y., Sherman, Y., and Ben-Sasson, S. A. (1992). Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 119, 493-501. Hall, D. H., Gu, G., Garcia-Anoveros, J., Gong, L., Chalfie, M., and Driscoll, M. (1997). Neuropathology of degenerative cell death in Caenorhabditiselegans. J. Neurosci. 17, 1033-1045. Hayat, M. A. (1970). Principles and Techniques of Electron Microscopy: Biological Applications, Volume 1 (New York, NY: Van Nostrand Reinhold Company). Hedgecock, E. M., Sulston, J. E., and Thomson, J. N. (1983). Mutations affecting programmed cell deaths in the nematode Caenorhabditiselegans. Science 220, 1277-1279. Hengartner, M. 0., Ellis, R. E., and Horvitz, H. R. (1992). Caenorhabditiselegans gene ced9 protects cells from programmed cell death. Nature 356, 494-499. 109 Hengartner, M. 0., and Horvitz, H. R. (1994). C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell 76, 665-676. Jacobson, M. D., Weil, M., and Raff, M. C. (1997). Programmed cell death in animal development. Cell 88, 347-354. Janicke, R. U., Sprengart, M. L., Wati, M. R., and Porter, A. G. (1998). Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 273, 9357-9360. Kaiser, N., and Edelman, I. S. (1977). Calcium dependence of glucocorticoid-induced lymphocytolysis. Proc. Natl. Acad. Sci. U. S. A. 74, 638-642. Kerr, J. F. R., Wyllie, A. H., and Currie, A. R. (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer. 26, 239257. Krause, M., and Hirsh, D. (1987). A trans-spliced leader sequence on actin mRNA in C. elegans. Cell 49, 753-761. Kroemer, G., Dallaporta, B., and Resche-Rigon, M. (1998). The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 60, 619-642. Kyte, J., and Doolittle, R. F. (1982). A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105-132. Lipton, S. A., and Nicotera, P. (1998). Calcium, free radicals and excitotoxins in neuronal apoptosis. Cell Calcium 23, 165-171. Martin, S. J., Reutelingsperger, C. P. M., McGahon, A. J., Rader, J. A., van Schie, R. C. A. A., LaFace, D. M., and Green, D. R. (1995). Early Redistribution of Plasma Membrane Phosphatidylserine Is a General Feature of Apoptosis Regardless of the Initiating Stimulus: Inhibition by Overexpression of Bcl-2 and Abl. J. Exp. Med. 182, 1545-1556. McConkey, D. J. (1996). The role of calcium in the regulation of apoptosis. Scanning Microsc. 10, 777-793. Mello, C. C., Kramer, J. M., Stinchcomb, D., and Ambros, V. (1991). Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959-3970. Metzstein, M. M., Stanfield, G. M., and Horvitz, H. R. (1998). Genetics of programmed cell death in C. elegans: Past, present and future. Trends Genet. 14, 410-416. Nagamine, K., Kudoh, J., Minoshima, S., Kawasaki, K., Asakawa, S., Ito, F., and Shimizu, N. (1998). Molecular cloning of a novel putative Ca 2 + channel protein (TRPC7) highly expressed in brain. Genomics 54, 124-131. 110 Nicotera, P., and Orrenius, S. (1998). The role of calcium in apoptosis. Cell Calcium 23, 173-180. Pettmann, B., and Henderson, C. E. (1998). Neuronal cell death. Neuron 20, 633-647. Porter, A. G., and Janicke, R. U. (1999). Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 6, 99-104. Putney, J. W., Jr., and McKay, R. R. (1999). Capacitative calcium entry channels. Bioessays 21, 38-46. Ren, Y., and Savill, J. (1998). Apoptosis: The importance of being eaten. Cell Death Differ. 5, 563-568. Riddle, D. L., Blumenthal, T., Meyer, B. J., and Priess, J. R. (1997). C. elegans II (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press). Robertson, A. M. G., and Thomson, J. N. (1982). Morphology of programmed cell death in the ventral nerve cord of Caenorhabditiselegans larvae. J. Embryol. exp. Morphol. 67, 89-100. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press). Saunders, J. W., Gasseling, M. T., and Saunders, L. C. (1962). Cellular death in morphogenesis of the avian wing. Dev. Biol. 139, 169-185. Scott, K., and Zuker, C. (1998). TRP, TRPL and trouble in photoreceptor cells. Curr. Opin. Neurobiol. 8, 383-388. Stroh, C., and Schulze-Osthoff, K. (1998). Death by a thousand cuts: an ever increasing list of caspase substrates. Cell Death Differ. 5, 997-1000. Sulston, J. E. (1976). Post-embryonic development in the ventral cord of Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 275, 287-297. Sulston, J. E., and Horvitz, H. R. (1977). Post-embryonic cell lineages of the nematode Caenorhabditiselegans. Dev. Biol. 56, 110-156. Sulston, J. E., Schierenberg, E., White, J. G., and Thomson, J. N. (1983). The embryonic cell lineage of the nematode Caenorhabditiselegans. Dev. Biol. 100, 64-119. Thompson, C. B. (1995). Apoptosis in the pathogenesis and treatment of disease. Science 267, 1456-1462. Thornberry, N. A., and Lazebnik, Y. (1998). Caspases: enemies within. Science 281, 13121316. Vaux, D. L., and Korsmeyer, S. J. (1999). Cell death in development. Cell 96, 245-254. 111 Wood, W. B., and the Community of C. elegans Researchers (1988). The Nematode Caenorhabditiselegans (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press). Wu, Y. C., and Horvitz, H. R. (1998). C. elegans phagocytosis and cell-migration protein CED-5 is similar to human DOCK180. Nature 392, 501-504. Wyllie, A. H., Kerr, J. F. R., and Currie, A. R. (1980). Cell death: the significance of apoptosis. Int. Rev. Cytol. 68, 251-306. 112 Table 1. Mutation of ced- 1 does not result in survival of cells that normally die. Genotype No. extra cellsa Range of extra cells wild type 0.04 ±0.03 0-1 ced-11(n2744) 0.09 ±0.06 0-1 ced-3(n2427) 1.0 ±0.1 0-4 ced-11(n2744); ced-3(n2427) 2.0 ± 0.2 0-5 ced-8(n1891) 0.7 ±0.1 0-3 ced-11(n2744); ced-8(n1891)b 1.4 ± 0.2 0-3 aAverage number of extra cells present in the anterior pharynx of L3-L4 hermaphrodites ± s.e.m. At least 23 animals of each strain were counted. bThis strain also contains the mutation lon-2(e678), which does not affect cell survival. Table 2. Mutations that prevent cell deaths are epistatic to ced-11. 3-fold embryosa 1.5-fold embryosa No. non-refractile structures Strain No. refractile corpses No. non-refractile structures nb No. refractile corpses ced-II(n2 744) 0.0 ± 0.0 5.5 ± 0.4 10 0.1 ± 0.07 0.5 ± 0.2 21 ced-5(n1812) ced-3(n717) NDC ND 0.0 ± 0.0 0.0 ± 0.0 20 ced-11(n2744); ced-5(n1812) ced-3(n717)d ND ND 0.1 ± 0.06 0.2 ± 0.08 30 ced-4(n]162)d 0.0± 0.0 0.0 ±0.0 18 ND ND ND ced-4(n1162) ced-11(n2744)d 0.0 ± 0.0 0.0 ± 0.0 18 ND ND ND nb aThe number of refractile corpses and non-refractile structures in the head of the appropriate stage embryos were counted. Errors indicate standard error of the mean. bn, number of animals examined. cND, not determined dThis strain contained the mutation dpy-17(e164), which does not affect programmed cell death. Figures Fig. 1. A visual screen for mutants with altered patterns of cell corpses in C. elegans embryos. One or two mutagenized L4 sem-4; ced-5 hermaphrodites was placed on a plate and allowed to grow and reproduce. F3 embryos inside F2 bags of worms were examined using Nomarski microscopy (Ellis et al., 1991) for any change from the Ced-5 pattern of cell corpses. Bags containing embryos with an altered cell-corpse pattern were recovered to seeded Petri plates. 115 Figure 1 EMS sem-4(n]378); ced-5(n2098) F1 F2 Look at F3 embryos inside F2 mothers by Nomarski microscopy: ced-5 bag of worms: progeny have many cell corpses ced-5; m/+ or n/rm: progeny have an altered pattern of cell corpses Fig. 2. Refractile cell corpses are reduced in number and abnormally non-refractile structures are present in ced-11; ced-5(n2098) embryos. (A) The number of refractile cell corpses in the head of 3-fold stage embryos of the indicated genotype. (B) The number of non-refractile, abnormal nuclei in the head of 3-fold stage embryos. Error bars indicate standard error of the mean. At least 28 embryos of each genotype were examined. 117 Figure 2 A 35 I 30250 20156 10- z 0- B ced-l1 ced-il ced-i (n2832); (n2833); (n2834); ced-5 ced-5 ced-5 ced-5 ced-li (n2744); ced-5 ced-I (n2745); ced-5 ced-5 ced-il (n2744); ced-5 ced-IlI ced-Ji ced-Ji (n2745); (n2832); (n2833); ced-5 ced-5 ced-5 . 35 300 25- a 200 150 C 10- z 0- ced-Ji (n2834); ced-5 Fig. 3. Ced-11 phenotype. (A) Nomarski micrograph of a ced-5(n2098) 3-fold stage embryo with refractile cell corpses. (B) ced-11(n2744); ced-5(n2098) 3-fold stage embryo with non-refractile cell corpses. (C) ced-11(n2744); ced-5(n2098) Li larva with nonrefractile cell corpses. (D) ced-11(n2744) 1.5-fold embryo with non-refractile cell corpses. Arrow, refractile cell corpse; arrowhead, non-refractile cell corpse. Scale bar indicates 10 pm. 119 -M Fig. 4. The engulfment of non-refractile ced-1 1 cell corpses is delayed by mutations that delay the engulfment of normal cell corpses. The y axis indicates the number of nonrefractile cell corpses present in the head of 3-fold stage ced-11(n2744); ced(engulfment) mutant embryos. Error bars, standard error of the mean. The full genotypes of the strains examined were ced-11(n2744), ced-1(e1735); ced-11(n2744), ced-11(n2744); ced-2(el752) dpy-9(e12), ced-I*(n2744); ced-5(n2098), ion-i(e185) ced-6(n1813) ced-11(n2744) and ced-11(n2744); and ced-10(n1993) dpy-20(e1282). 121 Figure 4 302520S15- 2 10Z 50- ced-11 ced-1; ced-11 ced-11; ced-11; ced-6 ced-5 ced-11 ced-2 ced-11; ced-10 Fig. 5. Kinetics of cell corpse appearance in ced-11 mutants. (A) Refractile cell corpse pattern in wild-type embryos. (B) Non-refractile cell corpse pattern in ced-11(n2744) embryos. (C) Refractile cell corpse pattern in ced-5(n2098) embryos. (D) Non-refractile cell corpse pattern in ced-11(n2744); ced-5(n2098) embryos. The numbers of refractile and non-refractile cell corpses in the head of embryos at different embryonic stages were counted using Nomarski microscopy (Ellis et al., 1991). Stages are: B/C, bean and comma; 1.5X, 1.5-fold embryos; 2X, 2-fold embryos; 3XE, early 3-fold embryos; 3XL, late 3-fold embryos with a well-developed pharynx and grinder. The y axis indicates the average number of cell corpses observed ± standard error of the mean. 123 Figure 5 A 45 40o 35- B J 45 U,40 wild type ced-11 o30- .,25- S2520- 20- 15- 150 Z 10- 6 10z 5- 5- 0- 0- B/C 1.5X 2X 3XE 40- B/C 3XL D C 45 ced-5 I 2X 45- 3XE 3XL ced-11; ced-5 35 30 - 9. 30- 1.5X 40 - 9. ) 35U I L 35 0 0'30- 6 - 025- 25- 820- 20 - 215- S15- Z 10- 10z 5- 0- 50- B/C 1.5X 2X 3XE 3XL B/C 1.5X 2X 3XE 3XL Fig. 6. Nomarski and electron micrographs of cell corpses. (A, B) Wild-type cell corpse. (C, D) ced-1 cell corpse. (E, F) ced-1; ced-11 cell corpses. In A, anterior is left; in C and E, anterior is right. In E, the posterior corpse is barely visible in this focal plane. Arrows, cell corpses in Nomarski micrographs. Arrowheads, boundary of plasma membrane of dying cell. Scale bar for Nomarski micrographs is as in Fig. 3. Scale bar for micrographs B and D indicates 500 nm. For micrograph F, 8 mm is approximately equivalent to 500 nm. 125 Fig. 7. ced-1 1 cloning. (A) ced-1 1 maps genetically near glp-1 (see Materials and Methods). (B) Cosmids assayed for transformation rescue. Brackets indicate cosmids injected together as pools with the fraction of transgenic lines showing rescue of the Ced-11 phenotype shown below. For singly injected cosmids, fraction of lines showing rescue is shown to the right of the cosmid name. (C) Subclones assayed for transformation rescue; fraction of lines showing rescue is indicated in the right-hand column. B, BamHI; H, HpaI; N, NheI; P, PstI. (D) A composite of the C. elegans ced-11 transcript derived from cDNA sequencing and 5' RACE analysis. The SL1 sequence is not shown. Intron positions are indicated by vertical bars. The predicted CED-11 protein sequence is shown beneath. Arrows designate base changes in the indicated ced-1 1 alleles. The termination codon is double-underlined. 127 Figure 7 A ON ryoI) ON -I--u 0.5 m.u. B glp-] ZC84 C15F7 (0/1) C28C8 T23G5 C37G7 (0/10) F02A9 T02C1 C03A8 (0/3) ZK507 (0/5) MO1A8 C33D7 (5/7) I- -I 5/15 0/4 8/14 C C33D7 -II 5 kb ZK512.3 Rescue? pGS10 pGS 16 pGS13 pGS 14 B B B P I I B B' H NN ' I B I 5/5 3/4 0/7 0/7 iii Figure 7 M D E T D P V P D M D L L P D T V P V V V S G D G S A A G I Q L C P A L I L G S P Q A L P G A I S L S T K Y M S S V E N G L K S F L I G E H w23 A Q A E V L H I L V N S D D M I A S D 48 300 98 450 148 S S I A K L R A A T A V K L A P P P 600 198 K H I Y S R L A A A A S E V D Q G V P D L I I S L I S H G N C G T W L I S S G E V N D P M S R V A S G A L K N V L P Q L T T N S K S V V D T S L N T L L L I C R K E P N D S Y E T V T A CT-AAC A AIL YGS IACAAIIGG'PCGTACAPTCG 2GGTTGTGTCCTGACCAATTAAl~ri"I900 F F V E H G I P V I I L Q D S C E L C A I L H S S H L L L E Q L I E F I D S S A Y P T S N M L I R M M H L S F G V D N L F 0 E P V A F S M V D G E E R S S V L Q L S P P I R V F A N D I Q K L V K K I V A K C L A A F A P T K L L V N S V N S A I D S L L G S G T LS SN P D P N V F C N D S S H R D K H E S I R I L A I W S L L L H L S R I A K S L G H K F Y F N TH T L A F H N A R E I I A H I C I S A V L I I P V K F W M L V R P R E S S R S E A L T A L T A P L S T A F ~2400 C G S N L W 0 T G M W V W S F F W W I E P I F L P T K Q D T V S P T V A L L D V G K F P Q K Q R A I S T L N I E E I 0 K C C L A T A W R V F A Y S V VL F A R T AR VL D S I Y S A K L F H I A L V V K I P L Q S H V TICAC~cAICIICAATA±~ R I V F S P I K W A K S V I D P R P P G S L V I LL F A F F V F L V S Y S T L F T Y I P L S D I F G P M I A V V AF P 0 R P V T M E V F R K T L S W A F F V L L S S A V I Y D V V MK H P S ~ N AT L V A F L HFL DL T n283d Pb8 P F P T D N T A I K L Y E A L C S Q A E r L S T K AC L I N S C Y G E R S Y L M D Y L W V M G K A V C R V V Q H G S C V K S L W N L V R V E I Q L S T C I L T C A Q F T 750 298 1050 T 348 1200 39 8 7 A n3745* Rh K 1350 R7 448 1500 R 498 1650 548 1800 598 E 1950 R 648 2100 698 G 2250 Y 748 ASATTFCIGTITAG2I13IATCGAAGAGAAITGTAWGAATACI S Q L Y P L T S N F D N D K F I S W LR F 0 P AGATGGrA7A0 248 LL H F S E SLI E L T KE S D T N GA FA A T F H H A S VAI FP KRP L S F D SE G C A A ALGPEHP C 150 S R E VLGI P VL FPVT K L M L A 24 ooa I WAh S L F T V V F T N F L R D T D L S L S K L L L E S E T C R K S F L G A P K R Y C S S V G Q Y A N P S C P S Q S L P A Y L I V I E Y F V I L W P I L F A F F S K T A K N V D D E A D K I W R F Q L Y S L A E 0 F R L R p p L P P P L T I F C L I C S A C C R F S N S F S G F F S D F D H P D F E A R D K C R T T W K F G S I Y R N P S V P F K R N E P A N S G T R K A S E L Q F F E K Y P E S ATTTGAIAAA n2833 D F N P HL P S N G E NS D R V Q T G S KDE K M Y A V G H D R L I G T A N S T H IL E P Q R N F V G KHMISGI G A N I G E N L K I S AAA L R K R SOD V C I Y G W R Y M N G R IRHTLRDSYE IHNH TQQKAKISANKSELQELHN FVNSFWRKLSHEQWKN V S V HH V S I R L K P W T V L V P R Y N P P F Y C K D V T S S N D K C W K L S A A G F P V D S Q N I P N E CAGTr Q S S A G N G V RR E A K C Q V L DA TCGIrATprIGcPIAGAoAGGAAAAACGAAAACGATAACAACGICGA0ZAGATATflTIGOTAT±I±I±I±CGAGAACArGATAATAACACAATTAClATIC01GGCICCIACGAIGCAAICG KRI K PS NODT AAAGCTIOGAAGAATAAAAA 3000 998 3150 1048 3300 1098 3450 1148 3600 1198 3750 1248 L A n2745* Wambser MU3900 1298 W E S A H L A S E V A E N E N D T D N A W T E H D V W A I S L R E R R I I T T I I G Y S W L P T S A I R wACAACITIATAAATTAAATI~CACAAAIGTATSITIUPIITITrAAACATAGCAAGATAAITAAT JAACAIGTICCA3CAA0CAGAC IGITPI±CGASAAAGCMTATAC~AoI G T V L P W Q A D L V F R A K T I Y P ~ 798 2550 848 2700 898 2850 948 4050 1348 4200 1398 4350 1418 4370 Fig. 8. ced-I1 transcript. (A) Northern blot of developmentally staged wild-type hermaphrodites probed with ced-1 1 (top) and reprobed with cyt-1 (Hengartner and Horvitz, 1994) (bottom) as a loading contrml. cyt-1 expression levels do not vary developmentally. The "young adult" lane contains RNA from a population of nongravid hermaphrodites. 130 Figure 8 5.0- 4.4- ced-11 cyt-1 Fig. 9. CED-11 protein. (A) Alignment of similar sequences of CED-11, human TRPC7 (Nagamine et al., 1998) and mouse melastatin (Duncan et al., 1998). Numbered horizontal bars indicate predicted transmembrane-spanning and pore-forming sequences in CED-11. (B) Kyte-Doolittle hydropathy plot of CED-11 (Kyte and Doolittle, 1982) (window size 11). Numbered bars indicate predicted transmembrane regions based on both hydrophobicity and similarity with other TRP-family proteins. (C) Comparison of the degree of similarity between the N-terminal, transmembrane and C-terminal domains of CED-11 and TRPC7 or melastatin. Numbers indicate percent similarity for each region or overall; numbers in parentheses indicate percent identity for each region or overall. 132 Figure 9 A MW7 TRP7 mtin F K LCGYTHEQHLEEATK 140 PHT PQGTQWDPKKHVQEMPTDAFGDIVFTGLSQKVK 0 a ffe nWI ME mostan - EE tpin Ga p ms A A I D i mK GPIM melastatin GN 7 It meatINp tath &Y LA moA-= - AW L ALPT A-L G K E K E E E N A T DM E -M-..AT E GP DGKE NAU2 Ni3f0--l=ypfii-~ P CEOl in9ES C m 7 MEPSALRKAGSEQEEGFEGLPRRVTDLGMVSNLRRSNSSLFKSWRLQCPFGNNDKQ ME EN LIL C - - - L - 295 KG-KEE---T- -.-.--- SFAF - TA-K-E- MN EMAC - - - - -Q ATL - - - -9 - 61 E-I 3 GEW77 II me atln - s- - ---- TR"7 - - - M~I S 76lMYHA 10 meatatin malastain M3E 15-- TRPC7 molastati CEO-11 tT7 -- - YE F. T P L R I - - - 1 A S30 W14 E 133 VW U - - - - - - -- - Figure 9 B 2[0 4.0 3.0 2.0 1.0 .5 0.0 0 -1.0 -2.0 -3.0 -.. - - -0--- -- 400 -n -n ---. -- - -- - 10 )0 800 600 - - - - ------ -- ----- 1400 1200 - ---- - - - - - - - - ---- .... .....- -4.0 I C II III IV V VI 743 I 50 1418 CED- 11 1 TRPC7 32% (15%) I 27 1 melastatin 32% (14%) I= 1102 30% (13%) 1 749 30%(13%) 1503 28% (15%) 59 1533 27%(13%) Fig. 10. Pathway model. In the C. elegans cell-death pathway, ced-11 functions downstream of genes required for cell killing and independently of genes required for cell-corpse engulfment. 135 Figure 10 living cell 0 Execution egi-1 ced-9 ced-4 ced-3 dead cell Corpse formation ced-11 cell corpse 0 Engulfment ced-J ced-2 ced-6 ced-5 ced-7 ced-10 Degradation engulfing cell Chapter 4 NUC-1, a C. elegans DNase II homolog, functions in an intermediate step in DNA degradation within cells dying by programmed cell death Yi-Chun Wu, Gillian M. Stanfield and H. Robert Horvitz To be submitted to Cell. I examined the kinetics of TUNEL staining during embryogenesis in wild-type and nuc-1 animals, cloned nuc-1 and isolated and analyzed the allele nuc-1(n3335). Yi-Chun Wu showed that TUNEL staining was dependent on programmed cell death and analyzed the effect of mutations in the engulfment genes on DNA degradation using both the SYTO 11 and TUNEL assays. We developed the TUNEL assay together. 137 Summary One hallmark of apoptosis is the degradation of chromosomal DNA. We cloned the C. elegans nuc-1 gene, which is involved in the degradation of the DNA of apoptotic cells, and found that nuc-1 encodes a homologue of the mammalian acidic endonuclease DNase II. We used the TUNEL (TdT-mediated dUTP nick-end labeling) technique to assay DNA degradation in nuc-1 and other mutants defective in apoptosis or programmed cell death and found that nuc-1 functions within dying cells and that the genes ced-1 and ced-7, which are involved in the engulfment of dying cells, also are involved in the generation of TUNEL-reactive DNA and may act upstream of nuc-1. DNA persists within unengulfed cell corpses in engulfment mutants, suggesting that the process of DNA degradation may require additional DNases provided by the engulfing cell. 138 Introduction Programmed cell death is important for the development of and homeostasis in metazoans and both the morphological changes occurring in dying cells and the molecular mechanisms by which cells die appear to be widely conserved (reviewed by Ellis et al., 1991b; Steller, 1995; Jacobson et al., 1997). One characteristic feature of programmed cell death (apoptosis) is the internucleosomal fragmentation of DNA into approximately 180 bp repeats, also referred to as DNA laddering (Wyllie, 1980). A number of candidate cell-death endonuclease activities have been characterized. CPAN/CAD (caspase-activated nuclease/ caspase-activated DNase) and DFF45/ICAD (_DNA fragmentation factor 45 kDa/ Inhibitor of CAD) together constitute a DNase regulated by caspase cleavage (Liu et al., 1997; Enari et al., 1998; Halenbeck et al., 1998; Sakahira et al., 1998). DFF45/ICAD mutant mice are defective in chromatin condensation and DNA degradation during the cell-death process, but immune system development is normal, suggesting that cells undergo programmed cell death even though cell-corpse DNA is not degraded normally (Zhang et al., 1998). Other candidate cell-death endonucleases include deoxyribonuclease I (DNase I) (Peitsch et al., 1992; Peitsch et al., 1993), deoxyribonuclease II (DNase II) (Barry and Eastman, 1992; Barry and Eastman, 1993), inducible-lymphocyte Ca 2*/Mg2*-dependent endonuclease (ILCME) (Khodarev and Ashwell, 1996) and cyclophilins (Montague et al., 1997). Despite this abundance of candidate endonucleases, the regulation of DNA degradation in dying cells in vivo is not well understood. The TUNEL (IdT-mediated dUTP nick-end labeling) technique (Gavrieli et al., 1992) has been applied to studies of programmed cell death in many organisms. Terminal deoxynucleotidyl transferase (TdT) is used to label DNA 3'-hydroxyl ends in situ with modified nucleotides detectable by fluorescence or immunohistochemistry. TUNEL specifically labels dying cells, since these cells have more free DNA ends than do viable cells as a consequence of DNA degradation (Wyllie, 1980; Oberhammer et al., 1993). Although TUNEL has been a diagnostic for cell corpses in many experimental models of programmed cell death, fundamental questions remain to be answered. For instance, are all intermediates during DNA degradation TUNEL-reactive, or does TUNEL label dying cells only during a specific stage of the death process? Time-course studies of DNA degradation in apoptotic mammalian epithelial cells using gel 139 electrophoresis suggest that DNA degradation involves multiple steps (Oberhammer et al., 1993). The chromosomal DNA of some apoptotic cells is cleaved at the chromatin loop domains, generating 50 kb fragments (Oberhammer et al., 1993). The subsequent cleavage of DNA at the internucleosomal linker region produces the characteristic DNA ladder (Wyllie, 1980; Oberhammer et al., 1993). Two findings suggest that the DNA fragments generated by DNA laddering may be those detected by TUNEL staining. First, in apoptotic lymphocytes the DNA fragments generated by DNA laddering contain 3-hydroxyl ends (Alnemri and Litwack, 1990) which should be substrates for TdT. Second, the appearance of TUNEL-positive signals correlates with the appearance of DNA laddering in dexamethasone-treated apoptotic thymocytes in culture (Gavrieli et al., 1992). Programmed cell death in the nematode Caenorhabditiselegans is morphologically and molecularly similar to that occurring in mammals (reviewed by Metzstein et al., 1998). In C. elegans, cells undergoing programmed cell death adopt a refractile, raised button-like appearance as observed by Nomarski differential interference contrast optics (Sulston and Horvitz, 1977). The time and place of death are known for each cell programmed to die and are essentially invariant from animal to animal. During the development of the hermaphrodite, 131 of the 1090 somatic cells generated undergo programmed cell death (Sulston and Horvitz, 1977; Sulston et al., 1983). Most (113/131) deaths occur during embryogenesis, many (111) during a short period between 220 and 470 minutes after fertilization (Sulston et al., 1983). The execution of cell deaths in C. elegans is controlled by the genes egl-1, ced-9, ced-4 and ced-3 (egl, egg-!aying defective; ced, cell death abnormal). Loss-of-function (lf) mutations in egl-1, ced-4, ced-3 or a gain-of-function (gf) mutation in ced-9 cause cells that normally die to survive. Each of these worm cell-death proteins is represented by one or more mammalian counterparts that also have been implicated in cell death: EGL-1, BH3-only Bcl-2-family proteins; CED-9, Bcl-2 family; CED-4, Apaf-1; and CED-3, caspases (reviewed by Metzstein et al., 1998). After cells die by programmed cell death, cell corpses are swiftly engulfed by neighboring cells (reviewed by Ellis et al., 1991b). Six genes, ced-1, ced-2, ced-5, ced-6, ced-7 and ced-10, important for cell-corpse engulfment have been characterized (Hedgecock et al., 1983; Ellis et al., 1991a). Mutations in any of these genes result in the persistence of many unengulfed cell corpses. In ced-1 and ced-2 mutants Feulgen140 reactive material is visible in the persistent cell corpses (Hedgecock et al., 1983), suggesting that DNA degradation does not proceed to completion. Thus, degradation of the DNA in cell corpses may require the ced-1 and ced-2 genes or an activity provided by engulfing cells. One C. elegans gene, nuc-1 (ujclease) is important for DNA degradation but does not appear to be involved in either the execution of cell death or the engulfment of cell corpses (Sulston, 1976). In nuc-1 mutants, both cell death and engulfment occur, but the pycnotic DNA of dead cells is not degraded and persists as a compact mass of Feulgenreactive material (Sulston, 1976; Hedgecock et al., 1983). The nuc-1 gene is also required to digest the DNA of the bacteria on which the animals feed: persistent bacterial DNA can be detected in the intestinal lumina of nuc-1 mutants, but not in those of wild-type animals (Sulston, 1976). The kinetics of DNA degradation in vivo can be studied in C. elegans by comparing TUNEL-staining patterns with the detailed knowledge of the cell-death pattern. In addition, by determining the TUNEL-staining patterns of C. elegans mutants defective in different aspects of programmed cell death, such as the execution of death and the engulfment of cell corpses, one can determine when DNA degradation occurs and how it may be regulated during the cell-death process. To analyze the control of DNA degradation during programmed cell death, we cloned the nuc-1 gene and assayed cellcorpse DNA degradation in the C. elegans wild-type embryo and in mutants defective in different aspects of the cell-death process. 141 Results TUNEL labels a subset of dying cells in C. elegans We developed a TUNEL protocol for C. elegans (see Experimental Procedures). We used this protocol to stain wild-type embryos and quantitated the number of TUNEL-postive nuclei. We chose to quantitate these nuclei in 1.5-fold stage embryos (approximately 420 minutes after fertilization) for two reasons. First, embryos progress through this easily recognizable stage in less than 20 minutes. Such a short time window should give highly reproducible TUNEL-staining patterns from embryo to embryo. Second, embryos at this stage contain an average of 14 refractile, dying cells, and prior to this stage 68 cells have already died and been engulfed (Sulston et al., 1983). Therefore, at this developmental stage we could monitor DNA degradation both in cells that are dying and cells that have died earlier and have been engulfed. We found that in labeled wild-type 1.5-fold embryos on average 1.7 nuclei were TUNEL-positive (Table 1; Figure 1A). TUNEL-positive signals colocalized with DNA as assayed by DAPI staining (data not shown). The refractile appearance of dying cells by Nomarski optics was not retained during fixation, so we could not identify cell corpses by their morphology in these fixed embryos. To confirm that TUNEL staining is specific to dying cells, we examined the TUNEL-staining patterns in the embryos of ced-9(nl950gf) (Hengartner et al., 1992), ced-4(n1162) and ced-3(n717) (Ellis and Horvitz, 1986) mutants, in which nearly all programmed cell deaths are blocked. We detected almost no TUNEL-positive nuclei in these mutants (Table 1), suggesting that the TUNEL-reactive nuclei in wild-type animals are those of dying cells. nuc-1 embryos have more TUNEL-reactive nuclei than do wild-type embryos In nuc-1 mutants, programmed cell death and cell-corpse engulfment still occur, but the DNA of dead cells is not completely degraded (Sulston, 1976; Hedgecock et al., 1983). We observed many more TUNEL-reactive nuclei in nuc-1 embryos than in wild-type embryos (Table 2; Figure 1B). Similar results were obtained using each of the three alleles of nuc-1, e1392, n334 and n887. All of these mutants contained the normal number of refractile corpses as compared to the wild type when observed using Nomarski microscopy (Table 2), suggesting that cell-death execution is not affected in these mutants. 142 To address whether the increase in TUNEL-reactive nuclei in nuc-1 embryos is specific to programmed cell death, we examined TUNEL-staining patterns of ced-9(nl950gf); nuc-1(e1392), ced-4(nl162); nuc-1(e1392) and ced-3(n717); nuc-1(e1392) embryos. These stained embryos had almost no TUNEL-positive signals (Table 2), showing that the presence of TUNEL-reactive DNA in nuc-1 embryos, just as in wildtype embryos, was dependent on the occurrence of programmed cell death. Since we observed only an average of 1.7 TUNEL-positive cells in wild-type embryos, it was possible that our TUNEL technique is not sensitive enough to detect all dying cells. However, it was also possible that DNA degradation might be a rapid process and that only certain transient intermediates are TUNEL-reactive during the process of cell death. The latter hypothesis is supported by"our finding that labeled nuc-1 embryos have many more TUNEL-positive signals than wild-type embryos. Time-course studies of the TUNEL-staining patterns in nuc-1 mutants (Figure 3 and data not shown) indicate that at least the majority of cell corpses become TUNELreactive at the appropriate time in nuc-1 mutants, suggesting that the initial step of DNA degradation by which cell-corpse DNA becomes TUNEL-reactive is not slowed in nuc-1 mutants. We hypothesize that nuc-1 is likely to mediate a subsequent step of the DNA degradation process involving either the masking of TUNEL-reactive DNA ends or the complete degradation of cell-corpse DNA. How long do nuclei persist in the TUNEL-reactive state in nuc-1 mutants? To address this question, we examined animals fixed and stained at later stages of embryogenesis. We observed many TUNEL-reactive nuclei in all 3.5-fold nuc-1 elongated embryos (data not shown). For example, in stained nuc-1(e1392) 3.5-fold embryos, we observed an average of 46 and a minimum of 30 TUNEL-positive cells. The 3.5-fold stage of embryogenesis corresponds to the period between approximately 520 minutes after fertilization and hatching at approximately 800 minutes; during this period, only two cells die. Our data for early embryonic stages suggest that mutations in nuc-1 do not delay significantly the onset of TUNEL reactivity; if cell corpses become TUNEL-reactive at the appropriate time in nuc-1 embryos, then many nuclei must remain TUNEL-reactive for at least several hours in nuc-1 animals. 143 nuc-1 encodes a DNase II homologue To understand the nature of the nuc-1 activity, we cloned the nuc-1 gene. Using genetic mapping, we localized nuc-1 to the interval between the cloned genes ced-8 and egl-15, and near or to the left of unc-115 (Figure 2A; see Experimental Procedures). We observed that a predicted gene, C07B5.5, encoding a protein with strong similarity to DNase II (Baker et al., 1998; Krieser and Eastman, 1998; Yasuda et al., 1998) lies within this region. The sequence of C07B5.5 is approximately 30% identical to that of human DNase II. Structural features are conserved between the C. elegans and human proteins: both proteins have a signal sequence, and the active site histidine is conserved (Baker et al., 1998; Krieser and Eastman, 1998; Yasuda et al., 1998). Since the biochemical characteristics of DNase II (e.g., Baker et al., 1998) are similar to those of the putative nuc-1 activity (Hevelone and Hartman, 1988), we sought to determine whether nuc-1 corresponds to C07B5.5. We first confirmed that the splicing pattern predicted for C07B5.5 by the C. elegans Genome Sequencing Consortium is correct by determining the sequences of cDNAs (generously provided by Yuji Kohara). We found that the cDNA yk434c9 contains the entire coding sequences of C07B5.5. We then determined the sequence of the C07B5.5 coding region and intron-exon junctions from the wild type and of the three nuc-1 alleles, e1392, n334 and n887. We found that all three mutants contain an identical change relative to the wild-type sequence, a G-to-A transition that is predicted to convert Trp59 to a TAG (amber) stop codon (Figure 2B). The nature of this lesion is consistent with genetic data that indicate that e1392 is amber-suppressible (Waterston and Brenner, 1978). That all three mutants contain the same base change is consistent with biochemical assays (Hevelone and Hartman, 1988) and our TUNEL assays, which revealed no phenotypic differences among these alleles. That all the existing nuc-1 alleles contain the same base change in the C07B5.5 gene raises the possibility that this lesion represents a commonly-occurring background mutation unrelated to the Nuc-1 phenotype. Therefore, we sought additional alleles of C07B5.5 using PCR to screen a library of mutagenized worms (Jansen et al., 1997) for animals carrying a deletion at the C07B5.5 locus. We identified the mutation n3335, a deletion of 2034 bp that extends from 363 bp before the predicted translation start of C07B5.5 to 166 bp before the start of exon 5 (Figure 2C). n3335 also is associated with a 2 bp insertion (sequence TA) at the deletion site. Since most of the C07B5.5 coding 144 region was eliminated in n3335, we expected that no functional protein should be produced. We examined n3335 animals to determine whether they are phenotypically Nuc. In nuc-1 mutant animals, pycnotic nuclei of dying cells persist (Sulston, 1976; Hedgecock et al., 1983). To test whether n3335 causes a Nuc phenotype, we scored for DNA persisting from the P lineage cell deaths, which occur during the Li and L2 stages in an approximately linear array along the ventral cord (Sulston and Horvitz, 1977). We tested the vital DNA-binding dye SYTO 11 (K.L. Hill and S.W. L'Hernault, personal communication) to determine whether its staining pattern appeared similar to that obtained using other DNA-labeling reagents. In L3, L4 and young adult wild-type animals, SYTO 11 labeled all nuclei. In SYTO 11-stained nuc-l(e1392) or nuc-1(n887) mutant animals, undegraded DNA of cell corpses appeared as additional bright, condensed foci of staining that did not coincide with nuclei visible by Nomarski microscopy. Undegraded bacterial DNA within the intestinal lumen also stained brightly. These patterns of staining were similar to those seen when wild-type and nuc-1 animals are stained with Feulgen (Sulston, 1976; Hedgecock et al., 1983) or DAPI (data not shown), indicating that SYTO 11 can be used as a vital stain for detecting the Nuc phenotype. The ventral cords of n3335 larvae contained extra SYTO 11-positive nuclei that appeared similar to those present in nuc-1(e1392) or nuc-1(n887) mutants (data not shown). Approximately one-quarter of the progeny of n3335/+ animals contained extra SYTO 11-positive nuclei, indicating that the effect of n3335 is recessive and not maternally-rescued. We also observed bright staining within the intestinal lumina of n3335 animals (data not shown). We then used TUNEL to stain n3335 embryos and found that the pattern of positive nuclei in n3335 embryos was indistinguishable from that caused by the nuc-1 allele e1392 (Figure 3). Therefore, n3335 causes a recessive Nuc phenotype like that of the known mutations in nuc-1. We tested whether the n3335 mutation genetically complements the nuc-1 allele e1392. We used SYTO 11 to stain animals trans-heterozygous for the two alleles and found that, like either single mutant, these animals contained extra SYTO 11-positive nuclei in the ventral cord (data not shown; see Experimental Procedures). These data show that n3335 is an allele of nuc-1 and confirm that nuc-1 corresponds to the DNase II-like gene C07B5.5. 145 nuc-1 may function within dying cells The observation that unengulfed cell corpses in ced-1 and ced-2 mutants contain Feulgen-reactive DNA (Hedgecock et al., 1983) suggests that the engulfment process is necessary for complete DNA degradation during programmed cell death in C. elegans. To determine whether nuc-1-mediated degradation of cell-corpse DNA is dependent on engulfment by neighboring cells, we examined TUNEL-staining patterns in engulfment-defective mutants. If engulfment were required for nuc-1-mediated DNA degradation (which eliminates TUNEL reactivity), then cell-corpse DNA should become TUNEL-reactive in engulfment-defective mutant embryos. Alternatively, if engulfment were not required for the nuc-1-mediated step of DNA degradation, then very few TUNEL-reactive cells should be present in engulfment-mutant embryos. We found that ced-2, ced-5, ced-6 and ced-10 mutant embryos, although they contained many unengulfed cell corpses at the 1.5-fold stage, had no more TUNELreactive nuclei than did wild-type embryos (Table 3). Furthermore, ced-2; nuc-1, ced-5; nuc-1, ced-6; nuc-1 and ced-10; nuc-1 embryos had the same number of TUNEL-reactive nuclei as did nuc-1 embryos (Table 3). We obtained equivalent results for multiple alleles of ced-2, ced-5 and ced-6 and from the single described allele of ced-10. These results indicate that the engulfment genes ced-2, ced-5, ced-6 and ced-10 are not required for the initial steps of DNA degradation during which TUNEL-reactive DNA ends are generated and degraded. We further conclude that both the activity generating TUNEL-reactive DNA ends and the nuc-1 activity act in dying cells rather than in engulfing cells, since the failure to engulf a cell did not prevent a cell corpse from becoming TUNEL-reactive or proceeding past that state in a nuc-1-dependent process. Furthermore, ced-2; nuc-1, ced-5; nuc-1, ced-6; nuc-1 and ced-10; nuc-1 embryos frequently displayed TUNEL staining in unengulfed cell corpses that had detached from embryos and had been shed into egg fluid (Figure 4). This observation confirms further that the generation of TUNEL-reactive DNA ends can occur in dying cells independently of engulfing cells. 146 Completion of DNA degradation during programmed cell death may require engulfing cells We stained ced-2, ced-5, ced-6 and ced-10 engulfment mutants using SYTO 11. We found that SYTO 11 labeled the DNA of both living and dying cells and that pycnotic nuclei of unengulfed cell corpses in all of these mutants were SYTO 11-positive (Figure 5 and data not shown). Feulgen staining of persistent cell corpses of ced-2 mutants had indicated previously that DNA degradation is incomplete in ced-2 animals (Hedgecock et al., 1983). That DNA is present in unengulfed cell corpses suggests that some step(s) of DNA degradation must occur in engulfing cells or be activated by the engulfment process. The engulfment genes ced-1 and ced-7 are involved in the initiation of DNA degradation When we stained ced-1 and ced-7 embryos, we found a reduction in TUNEL-reactive nuclei in both of these mutants as compared to the wild type. ced-1 embryos had very few TUNEL-staining nuclei in either a wild-type or nuc-1 background (Table 4). Mutations in ced-7 reduced the number of TUNEL-reactive nuclei to a lesser degree: ced-7 embryos had fewer TUNEL-reactive nuclei than did wild-type embryos, and ced-7; nuc-1 embryos had fewer than half the TUNEL-reactive nuclei of nuc-1 embryos (Table 4). We observed similar staining patterns using multiple alleles of ced-1 and ced-7. The reduction in TUNEL staining did not correlate with the strength of the engulfment defect. The reduction of TUNEL signals in ced-1 and ced-7 mutants was not caused by the disappearance of DNA in cell corpses, since unengulfed cell corpses in these mutants stain with SYTO 11 (data not shown). Also, cell corpses in ced-1 mutants have been shown previously to contain Feulgen-reactive DNA (Hedgecock et al., 1983). Therefore, TUNEL staining may be reduced in ced-1 and ced-7 mutants because the degradation of cell-corpse DNA is blocked prior to the generation of TUNEL-reactive ends. These data suggest that ced-1 and ced-7 are involved in the generation of TUNEL-reactive DNA ends within dying cells during programmed cell death. 147 Discussion Endonuclease activity in dying cells We have shown that the gene nuc-1, which is important for the process of DNA degradation during programmed cell death in C. elegans, encodes a protein similar to mammalian DNase II. Previous results had indicated that an endonuclease activity detectable in wild-type C. elegans protein extracts is greatly reduced in or absent from protein extracts from nuc-1 mutants (Sulston, 1976; Hedgecock et al., 1983; Hevelone and Hartman, 1988). This endonuclease activity resembles that of mammalian DNase II in that both are independent of Ca2" and Mg2" and they have a similar acidic pH optimum (Hevelone and Hartman, 1988; Barry and Eastman, 1992). DNase II previously was implicated in DNA degradation during programmed cell death (Barry and Eastman, 1992; Barry and Eastman, 1993; Torriglia et al., 1995; Krieser and Eastman, 1998). For instance, it can induce DNA laddering in isolated nuclei (Barry and Eastman, 1993) and it has been localized to the nuclei of lens fibers undergoing an apoptosis-like death (Torriglia et al., 1995). However, many endonucleases have been suggested to function in apoptotic cells (Hughes and Cidlowski, 1994). Our finding that nuc-1 encodes a DNase II-like protein represents in vivo evidence that DNase II is important during programmed cell death. We adapted the TUNEL technique, which labels dying cells, for use in C. elegans, and we used this technique to determine that nuc-1 as well as the engulfment genes ced-1 and ced-7 act within the dying cells and the engulfment genes ced-2, ced-5, ced-6 and ced-10 act within the engulfing cells. We found that most (47/82) of the nuclei of dead cells in nuc-1 embryos were TUNEL-reactive, while in wild-type embryos only a small subset of dying cells stained by TUNEL, suggesting that DNA degradation is normally a rapid process and that only certain transient intermediates are TUNELreactive. Our data suggest that nuc-1 is not required for the initial step of DNA degradation by which TUNEL-reactive DNA is generated but rather for the generation of a subsequent TUNEL-unreactive intermediate in the degradation process. Under reaction conditions used for TUNEL assays, the TdT polymerase extends 3'-hydroxyl ends of single-stranded or double-stranded DNA molecules with blunt, recessed or overhanging ends (Roychoudhury et al., 1976). The cleavage of DNA by mammalian DNase II generates 5'-hydroxyl and 3'-phosphate ends, neither of which is a substrate 148 for TdT, consistent with our suggestion that nuc-1 generates DNA that is not TUNELreactive. However, it is not clear how a DNase II-like activity functions to eliminate TUNEL-reactive DNA ends. The activity that destroys or masks TUNEL-reactive DNA ends either may be dependent on nuc-1 or may act downstream of nuc-1. Regulation of NUC-1 nuc-1 is important not only for the degradation of the DNA of dying cells but also for degradation of DNA of bacteria (ingested by the worm as food) in the intestine. While nuc-1 appears to act within dying cells, it is presumably secreted into the intestinal lumen to act upon bacterial DNA. How might nuc-1 be regulated to accomplish these rather different tasks? Mammalian DNase II has been reported to be secreted into the medium when expressed in some mammalian cell types (Baker et al., 1998). NUC-1, like mammalian DNase II, has a predicted signal sequence at its N-terminus, suggesting that it, too, might be secreted by some cells. How might the activity of nuc-1 be regulated within a dying cell? The nuc-1dependent enzyme has an acidic pH optimum, so it is not expected to be active at the physiological pH of living cells. Cells undergoing programmed cell death may undergo intracellular acidification and thereby activate NUC-1. Such acidification has been demonstrated for some mammalian cell types (Barry and Eastman, 1992), but not yet for C. elegans programmed cell deaths. The NUC-1 activity also might be regulated at the level of either protein processing or subcellular localization. The maturation of human DNase II involves cleavage of the proprotein to produce fragments of approximately 30 kDa and 10 kDa (Baker et al., 1998; Krieser and Eastman, 1998; Yasuda et al., 1998). While the larger subunit appears to have endonuclease activity on its own [Krieser, 1998 #512, a biochemically-purified form of DNase II has been found to be a heterodimer of 35 kDa and 10 kDa peptides (Liao, 1985; Barry and Eastman, 1993), which might correspond to the two cleavage products of pro-DNase II. Intriguingly, the cleavage site of human DNase II lies after an aspartate residue (Baker et al., 1998; Krieser and Eastman, 1998; Yasuda et al., 1998), raising the possibility that a caspase might directly cleave and thereby activate DNase II in dying cells. However, the cleavage site is not obviously conserved between the human and C. elegans proteins, and the surrounding amino acid sequence (HEDD/S) does not conform to the consensus cleavage site for caspases in general or CED-3 in 149 particular (Xue et al., 1996; Talanian et al., 1997; Thornberry et al., 1997). Also, if cleavage by CED-3 were required for NUC-1 processing, then one might expect ced-3 mutants to have persistent bacterial DNA in the intestinal lumen, but they do not (Ellis and Horvitz, 1986; our unpublished observations). Additional caspase genes in C. elegans have recently been identified and shown to encode functional proteases (Shaham, 1998), so it is possible that one or more of these caspases, rather than CED-3, cleaves NUC-1. However, we think it more likely that another protease processes NUC-1. Whether this processing can depend upon the activation of programmed cell death remains to be determined. Additional activities required for DNA degradation Our results suggest that DNA degradation during programmed cell death involves at least three distinct steps (Figure 6). During the first step, TUNEL-reactive DNA ends are generated. The genes ced-1 and ced-7 must function for this process to occur normally, since mutations in either gene reduce the number of TUNEL-staining signals. The second step involves the nuc-1 gene, which mediates a conversion of TUNELreactive DNA into TUNEL-unreactive DNA. The first two steps occur in dying cells rather than in engulfing cells, since the engulfment process is not required for either the generation of TUNEL-staining DNA or progression to the TUNEL-unreactive state. ced-1, ced-7 and nuc-1 all appear to act in dying cells to promote DNA degradation. The third step involves the final degradation of TUNEL-unreactive DNA fragments and occurs in engulfing cells or is activated by the engulfment process. How are TUNEL-reactive DNA ends generated? Since mutations in ced-1 and ced-7 reduce the number of TUNEL-staining signals, these two genes could act in the generation of TUNEL-reactive DNA. However, we cannot rule out the possibility that mutations in these genes somehow render cell corpses insensitive to TUNEL reagents and result in decreased TUNEL staining. The nature of the ced-1 gene product has not been reported. The ced-7 gene encodes a protein similar to ABC transporters (Wu and Horvitz, 1998a). The CED-7 protein is localized to the plasma membrane (as visualized by immunostaining with anti-CED-7 antibodies) (Wu and Horvitz, 1998a), so CED-7 is unlikely to be directly involved in DNA degradation in the nucleus. The identification and characterization of substrate(s) transported by CED-7 should elucidate why this protein is important for both engulfment and DNA degradation. Mutations in ced-7 150 reduce the number of TUNEL-reactive nuclei but do not completely block the generation of TUNEL-reactive DNA. The n2094 and n1996 alleles are likely to be null, based on the nature of the molecular lesions in these mutants (Wu and Horvitz, 1998a). Therefore, the higher level of TUNEL staining in ced-7 embryos relative to that in ced-1 embryos probably is not a result of residual ced-7 activity. One possibility is that ced-7 acts with another gene or genes in a partially redundant way to regulate the generation of TUNEL-reactive DNA ends during programmed cell death, perhaps by activating an endonuclease. Finally, at least two other genes encoding DNase II-like proteins are present in the C. elegans genome (Krieser and Eastman, 1998). It is curious, therefore, that mutations in the nuc-1 gene cause such a profound effect on DNA degradation. It is possible that the other DNase II-like genes do not function in programmed cell death or at least play much less important roles. That DNase II-like activity is nearly absent from protein extracts in nuc-1 mutants (Hevelone and Hartman, 1988) indicates that the other two DNase Il-like genes may be expressed at very low levels, if at all. However, DNA of cell corpses does become TUNEL-unreactive eventually in nuc-1 mutants, although the process is very slow. Perhaps residual endonuclease activity provided by the other DNase II-like genes degrades the DNA. Alternatively, the TUNEL-unreactive state generated by nuc-1 activity may not be an obligatory intermediate in the DNA degradation pathway. Our cloning of nuc-1 should open many genetic and biochemical avenues for continuing to analyze the DNA degradation process during programmed cell death. Experimental Procedures Nematodes All strains were grown at 20'C on Petri dishes containing NGM agar seeded with E. coli OP50 bacteria (Brenner, 1974). The Bristol N2 strain (Brenner, 1974) was used as the wild-type strain. Alleles used are: LGI: ced-1(el735, n1995). LGIII: ced-6(n1813, n2095); ced-7(n1892, ni996, n2094); ced-4(nl162); ced-9(nl950). LGIV: ced-2(e1752, n1994); ced-5(n1812, n2691); ced-10(n1993), ced-3(n717). LGX: nuc-1(e1392, n334, n887, n3335); dpy-6(e14); unc-9(el0l); unc-10(e102); ced-8(nl891); lin-2(e1309); sma-5(n678); unc-27(e155); egl-15(n484); unc-115(e2225). All mutations except ced-5(n2691), ced-1(n1995) and nuc-1(n3335) are described by Riddle et al. (1997); ced-5(n2691) is described by Wu and 151 Horvitz (1998b), ced-1(n1995) is described by Ellis et al. (1991a) and nuc-1(n3335) is described in this study. Genetic mapping nuc-1 was mapped previously to linkage group X between the genes dpy-7 and unc-9 (V. Ambros and H.R.H., unpublished results). We used three-factor mapping to further localize the nuc-1 gene. To score the nuc-1 phenotype, we obtained homozygous lines from recombinant animals and scored for persistent DNA of engulfed cell corpses in the ventral nerve cords of L3 and L4 larvae stained with SYTO 11 (K.L. Hill and S.W. L'Hernault, personal communication). We mapped nuc-1 between dpy-6 and unc-9: from dpy-6 unc-9/nuc-1(e1392) mothers, 7/8 Unc non-Dpy and 2/8 Dpy non-Unc lines were Nuc, and from dpy-6 unc-9/nuc-1(n887) mothers, 7/8 Unc non-Dpy and 0/6 Dpy non-Unc lines were Nuc. We mapped nuc-1 between ced-8 and lin-2: from unc-10 ced-8/nuc-1(e1392) lin-2 mothers, 8/12 Unc Lin lines were Ced-8 and 2/8 of these Ced-8 lines were Nuc. We mapped nuc-1 left or near to the right of sma-5: from sma-5 lin-2/nuc-1(e1392) mothers, 4/4 Lin non-Sma lines were Nuc, and from sma-5 lin-2/nuc-1(n887) mothers, 11/11 Lin non-Sma lines were Nuc. We mapped nuc-1 between unc-27 and egl-15: from unc-27 egl-15/nuc-1(e1392) mothers, 9/15 Egl non-Unc and 3/10 Unc non-Egl lines were Nuc. We mapped nuc-1 to the left or very near right of unc-115: from unc-115 egl-15/nuc-1(e1392) mothers, 13/13 Egl non-Unc and 0/14 Unc non-Egl lines were Nuc. Complementation tests n3335 males were mated to unc-27(e155) nuc-1(e1392) egl-15(n484) hermaphrodites, and non-Unc progeny at the L3 or older stage were stained with SYTO 11 in a microtiter well, as described below. 50/50 animals examined were Nuc (had extra staining nuclei in the ventral cord). The presence of the n3335 deletion was confirmed by PCR for 20 of these animals. Cell-corpse counts The number of refractile cell corpses in living embryos was counted using Nomarski optics (Ellis et al., 1991a). 152 TUNEL Animals were washed off of one to three 100x15 mm Petri plates with water and treated with hypochlorite to obtain embryos (Wood and Researchers, 1988). Embryos were fixed using a modified protocol derived from Finney and Ruvkun (1990). 1 ml of fixation solution containing 80 mM KCl, 20 mM NaCl, 1.3 mM EGTA, 3.2 mM spermine, 7.5 mM sodium HEPES pH 6.5, 25% methanol, 2% paraformaldehyde and 0.4% glutaraldehyde was added to hypochlorite-treated embryos, which were then immediately frozen in liquid nitrogen. The frozen embryos were thawed in a water bath at room temperature for 2 min and rocked at room temperature for 25 min. Fixed embryos were then washed once in 1 ml Tris-Triton buffer (1% Triton X-100 and 100 mM Tris, pH 7.4) and three times in 1 ml PBST (IX PBS containing 0.5% Triton X-100) for ten min each. Approximately 3 ptl of packed embryos were preincubated with 25 g1 of TdT reaction buffer (200 mM sodium cacodylate, 25 mM Tris-HCl, 0.25 mg/ml bovine serum albumin, 0.1% Triton X-100, 1.5 mM cobalt chloride, pH 6.6) for 5 min at room temperature. The TdT reaction buffer was then replaced with TdT reaction buffer containing 0.2 unit of TdT (Boehringer Mannheim), 6.6 nM dUTP (Boehringer Mannheim) and 3.3 nM fluorescein-11-dUTP (Boehringer Mannheim) and incubated for 2 hr at 37 0 C. After incubation, embryos were washed in PBST three times for 10 min. For DAPI staining, fixed and stained embryos were incubated in 1 pg/ml DAPI in PBST for 10 min. Embryos were then mounted in VECTORSHIELD mounting medium (Vector Laboratories) and visualized using either a Zeiss Axiophot microscope equipped for fluorescence microscopy or a Bio-Rad MRC-500 confocal microscope. SYTO 11 staining Mixed-staged worms were washed off of plates with M9 buffer (Wood and Researchers, 1988), collected in a 1.5 ml Eppendorf tube, and washed once in M9 buffer. The washed worms were then rocked at room temperature for 1.5 hr in 1 ml of 10 nM SYTO 11 (Molecular Probes) in M9 buffer. The stained worms were then washed once in M9 and transferred to a Petri plate seeded with OP50 to recover for 30 min to 2 hr. Alternatively, worms were picked into 50 pl of M9 buffer in a microtiter well, 50 pI of 20 nM SYTO 11 was added and the worms were incubated at room temperature for 1.5 hr. Worms were mounted on 4% agar pads with 20 mM sodium azide and visualized using an FITC filter set. 153 Molecular biology Standard molecular biology methods were followed (Sambrook et al., 1989). The sequences of primers used for PCR amplification, DNA sequence determination and 5' RACE are available upon request. We isolated RNA from a population of mixed-stage N2 animals using standard methods and performed poly(A) selection using Fast Track (Invitrogen). 5'-RACE reactions were performed using the 5' RACE System 2.0 (Gibco). Deletion library screening We used PCR to screen a library of chemically mutagenized N2 nematodes and select animals containing a deletion in C07B5.5 essentially as described in Jansen et al. (1997). The deletion library was constructed by the members of the Horvitz laboratory as directed by R. Ranganathan and P. Reddien. 154 Acknowledgments We are indebted to R. Ranganathan and P. Reddien for engineering the lab C. elegans deletion library and for providing screening advice and moral support. We thank B. James for deletion library screening and for DNA sequence determinations, N. Tsung for assistance with nuc-1 mapping and Y. Kohara for nuc-1 cDNAs. We also thank B. Hersh, S. Cameron and M. Metzstein for comments concerning this manuscript and its less-evolved versions. G.M.S. was supported by a Howard Hughes Medical Institute Predoctoral Fellowship. H.R.H. is an Investigator of the Howard Hughes Medical Institute. 155 References Alnemri, E. S., and Litwack, G. (1990). Activation of internucleosomal DNA cleavage in human CEM lymphocytes by glucocorticoid and novobiocin. Evidence for a non-Ca 2 +requiring mechanism(s). J. Biol. Chem. 265, 17323-17333. Baker, K. P., Baron, W. F., Henzel, W. J., and Spencer, S. A. (1998). Molecular cloning and characterization of human and murine DNase II. Gene 215, 281-289. Barry, M. A., and Eastman, A. (1992). Endonuclease activation during apoptosis: the role of cytosolic Ca 2 + and pH. Biochem. Biophys. Res. Commun. 186, 782-789. Barry, M. A., and Eastman, A. (1993). Identification of deoxyribonuclease II as an endonuclease involved in apoptosis. Arch. Biochem. Biophys. 300, 440-450. Brenner, S. (1974). The genetics of Caenorhabditiselegans. Genetics 77, 71-94. Ellis, H. M., and Horvitz, H. R. (1986). Genetic control of programmed cell death in the nematode C. elegans. Cell 44, 817-829. Ellis, R. E., Jacobson, D. M., and Horvitz, H. R. (1991a). Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditiselegans. Genetics 129, 79-94. Ellis, R. E., Yuan, J. Y., and Horvitz, H. R. (1991b). Mechanisms and functions of cell death. Annu. Rev. Cell Biol. 7, 663-698. Enari, M., Sakahira, H., Yokoyama, H., Okawa, K., Iwamatsu, A., and Nagata, S. (1998). A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391, 43-50. Finney, M., and Ruvkun, G. (1990). The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell 63, 895-905. Gavrieli, Y., Sherman, Y., and Ben-Sasson, S. A. (1992). Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 119, 493-501. Halenbeck, R., MacDonald, H., Roulston, A., Chen, T. T., Conroy, L., and Williams, L. T. (1998). CPAN, a human nuclease regulated by the caspase-sensitive inhibitor DFF45. Curr. Biol. 8, 537-540. Hedgecock, E. M., Sulston, J. E., and Thomson, J. N. (1983). Mutations affecting programmed cell deaths in the nematode Caenorhabditiselegans. Science 220, 1277-1279. Hengartner, M. 0., Ellis, R. E., and Horvitz, H. R. (1992). Caenorhabditiselegans gene ced9 protects cells from programmed cell death. Nature 356, 494-499. 156 Hevelone, J., and Hartman, P. S. (1988). An endonuclease from Caenorhabditiselegans: partial purification and characterization. Biochem. Genet. 26, 447-461. Hughes, F. M., and Cidlowski, J. A. (1994). Apoptotic DNA degradation: evidence for novel enzymes. Cell Death Differ. 1, 11-17. Jacobson, M. D., Weil, M., and Raff, M. C. (1997). Programmed cell death in animal development. Cell 88, 347-354. Jansen, G., Hazendonk, E., Thijssen, K. L., and Plasterk, R. H. (1997). Reverse genetics by chemical mutagenesis in Caenorhabditiselegans. Nat Genet 17, 119-121. Khodarev, N. N., and Ashwell, J. D. (1996). An inducible lymphocyte nuclear Ca 2 +/Mg2 +-dependent endonuclease associated with apoptosis. J. Immunol. 156, 922931. Krieser, R. J., and Eastman, A. (1998). The cloning and expression of human deoxyribonuclease II. A possible role in apoptosis. J. Biol. Chem. 273, 30909-30914. Liao, T. H. (1985). The subunit structure and active site sequence of porcine spleen deoxyribonuclease. J. Biol. Chem. 260, 10708-10713. Liu, X., Zou, H., Slaughter, C., and Wang, X. (1997). DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 89, 175-184. Metzstein, M. M., Stanfield, G. M., and Horvitz, H. R. (1998). Genetics of programmed cell death in C. elegans: Past, present and future. Trends Genet. 14, 410-416. Montague, J. W., Hughes, F. M., Jr., and Cidlowski, J. A. (1997). Native recombinant cyclophilins A, B, and C degrade DNA independently of peptidylprolyl cis-transisomerase activity. Potential roles of cyclophilins in apoptosis. J. Biol. Chem. 272, 66776684. Oberhammer, F., Wilson, J. W., Dive, C., Morris, I. D., Hickman, J. A., Wakeling, A. E., Walker, P. R., and Sikorska, M. (1993). Apoptotic death in epithelial cells: cleavage of DNA to 300 and/or 50 kb fragments prior to or in the absence of internucleosomal fragmentation. EMBO J. 12, 3679-3684. Peitsch, M. C., Hesterkamp, T., Polzar, B., Mannherz, H. G., and Tschopp, J. (1992). Functional characterisation of serum DNase I in MRL-lpr/lpr mice. Biochem. Biophys. Res. Commun. 186, 739-745. Peitsch, M. C., Polzar, B., Stephan, H., Crompton, T., MacDonald, H. R., Mannherz, H. G., and Tschopp, J. (1993). Characterization of the endogenous deoxyribonuclease involved in nuclear DNA degradation during apoptosis (programmed cell death). EMBO J. 12, 371-377. 157 Riddle, D. L., Blumenthal, T., Meyer, B. J., and Priess, J. R. (1997). C. elegans II (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press). Roychoudhury, R., Jay, E., and Wu, R. (1976). Terminal labeling and addition of homopolymer tracts to duplex DNA fragments by terminal deoxynucleotidyl transferase. Nucleic Acids Res. 3, 101-116. Sakahira, H., Enari, M., and Nagata, S. (1998). Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 391, 96-99. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press). Shaham, S. (1998). Identification of multiple Caenorhabditiselegans caspases and their potential roles in proteolytic cascades. J. Biol. Chem. 273, 35109-35117. Steller, H. (1995). Mechanisms and genes of cellular suicide. Science 267, 1445-1449. Sulston, J. E. (1976). Post-embryonic development in the ventral cord of Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 275, 287-297. Sulston, J. E., and Horvitz, H. R. (1977). Post-embryonic cell lineages of the nematode Caenorhabditiselegans. Dev. Biol. 56, 110-156. Sulston, J. E., Schierenberg, E., White, J. G., and Thomson, J. N. (1983). The embryonic cell lineage of the nematode Caenorhabditiselegans. Dev. Biol. 100, 64-119. Talanian, R. V., Quinlan, C., Trautz, S., Hackett, M. C., Mankovich, J. A., Banach, D., Ghayur, T., Brady, K. D., and Wong, W. W. (1997). Substrate specificities of caspase family proteases. J. Biol. Chem. 272, 9677-9682. Thornberry, N. A., Rano, T. A., Peterson, E. P., Rasper, D. M., Timkey, T., Garcia-Calvo, M., Houtzager, V. M., Nordstrom, P. A., Roy, S., Vaillancourt, J. P., Chapman, K. T., and Nicholson, D. W. (1997). A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 272, 17907-17911. Torriglia, A., Chaudun, E., Chany-Fournier, F., Jeanny, J. C., Courtois, Y., and Counis, M. F. (1995). Involvement of DNase II in nuclear degeneration during lens cell differentiation. J. Biol. Chem. 270, 28579-28585. Waterston, R. H., and Brenner, S. (1978). A suppressor mutation in the nematode acting on specific alleles of many genes. Nature 275, 715-719. Wood, W. B., and the Community of C. elegans Researchers (1988). The Nematode Caenorhabditiselegans (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press). 158 Wu, Y. C., and Horvitz, H. R. (1998a). The C. elegans cell corpse engulfment gene ced-7 encodes a protein similar to ABC transporters. Cell 93, 951-960. Wu, Y. C., and Horvitz, H. R. (1998b). C. elegans phagocytosis and cell-migration protein CED-5 is similar to human DOCK180. Nature 392, 501-504. Wyllie, A. H. (1980). Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature 284, 555-556. Xue, D., Shaham, S., and Horvitz, H. R. (1996). The Caenorhabditiselegans cell-death protein CED-3 is a cysteine protease with substrate specificities similar to those of the human CPP32 protease. Genes Dev. 10, 1073-1083. Yasuda, T., Takeshita, H., lida, R., Nakajima, T., Hosomi, 0., Nakashima, Y., and Kishi, K. (1998). Molecular cloning of the cDNA encoding human deoxyribonuclease II. J. Biol. Chem. 273, 2610-2616. Zhang, J., Liu, X., Scherer, D. C., van Kaer, L., Wang, X., and Xu, M. (1998). Resistance to DNA fragmentation and chromatin condensation in mice lacking the DNA fragmentation factor 45. Proc. Natl. Acad. Sci. U. S. A. 95, 12480-12485. 159 Table 1. TUNEL specifically labels dying cells No. TUNELpositive cellsa Range of TUNELpositive cells No. cell corpsesb wild type 1.7 ±1.3 0-4 14± 1 ced-3(n717) 0.1 ± 0.2 0-1 0.0 ± 0.0 ced-4(n1162) 0.0 ±0.0 0-1 0.0 ±0.2 ced-9(n1950) 0.1 ±0.4 0-1 0.1 ±0.3 Genotype aTUNEL-positive nuclei in at least 60 embryos of each genotype were counted. bCell corpses in 15 wild-type embryos and in more than 50 embryos of each mutant genotype were counted using Nomarski optics. Embryos were scored at the 1.5-fold stage. The data shown are means ± s.e.m. Table 2. TUNEL labels persistent DNA in nuc-1 animalsa Genotype No. TUNELpositive cells (n=45) Range of TUNELpositive cells No. cell corpses (n=15) nuc-1(e1392) 47.8 ±4.8 38-64 14 ± 2 nuc-1(n334) 45.3 ±6.2 36-60 14± 2 nuc-1(n887) 47.5 ±5.3 35-59 14± 1 ced-3(n 717); nuc-1(e 1392) 0.4 ± 0.7 0-2 0.0 ± 0.0 ced-4(n 1162); nuc-1(e 1392) 0.2 ± 0.6 0-2 0.1 ± 0.2 ced-9(n 1950); nuc-1(e 1392) 4.2 ± 1.8 0-9 0.1 ± 0.4 aEmbryos were scored at the 1.5-fold stage. The data shown are means + s.e.m. Table 3. Mutations in ced-2, 5, 6 and 10 do not alter TUNEL staininga No. TUNELpositive cells (n=45) Range of TUNELpositive cells No. cell corpses (n=15) wild type 1.7 ±1.3 0-4 14±1 ced-2(e1752) 1.5 ±1.1 0-3 21± 3 ced-2(hn1994) 1.7± 1.4 0-3 20± 3 ced-5(hn1812) 1.5 ±1.0 0-4 39± 5 ced-5(n2691) 1.4± 1.2 0-3 34± 4 ced-6(n2095) 1.4± 0.9 0-3 29± 4 ced-6(hn1813) 1.6 ±1.4 0-3 33± 5 ced-10(n1993) 1.4 ±1.3 0-3 31± 4 nuc-1(e1392) 47.8 ±4.8 38-64 14± 2 ced-2(e 1752); nuc-1(e 1392) 44.6 ± 4.6 37-51 23 ± 3 ced-2(n 1994); nuc-1(e 1392) 47.6 ± 4.9 41-56 21 ± 3 ced-5(n1812); nuc-1(e1392) 45.1 ±5.8 39-59 38± 5 ced-5(n269 1); nuc-1(e 1392) 44.6 ± 4.6 35-56 35 ± 4 ced-6(n2095); nuc-1(e 1392) 46.2 ± 4.5 33-54 30 ± 5 ced-6(n 1813); nuc-1(e 1392) 43.6 ± 4.6 35-52 32 ± 5 ced-1O(n 1993); nuc-1(e 1392) 42.6 ± 4.3 37-51 33 ± 4 Genotype aEmbryos were scored at the 1.5-fold stage. The data shown are means s.e.m. Table 4. Mutations in ced-1 and ced-7reduce TUNEL staininga No. TUNELpositive cells (n=45) Range of TUNELpositive cells No. cell corpses (n=15) wild type 1.7 ±1.3 0-4 14± 1 ced-1(e1735) 0.5 ±0.7 0-3 24± 3 ced-1(n1995) 0.3 ±0.2 0-2 25± 4 ced-7(n2094) 0.7 ±0.8 0-3 34± 3 ced-7(n 1892) 0.5 ±0.4 0-2 35± 5 nuc-1(e1392) 47.8 ±4.8 38-64 14± 2 ced-1(e1735); nuc-1(e1392) 1.0 ±1.0 0-3 26± 3 ced-1(n1995); nuc-1(e1392) 1.2 ± 1.0 0-5 27± 5 ced-7(n2094); nuc-1(e 1392) 21.1 ± 1.3 15-28 38 ± 3 ced-7(n 1892); nuc-1(e 1392) 22.3 ± 3.2 14-29 37 ± 5 ced-7(n 1996); nuc-1(e 1392) 19.0 ± 1.7 16-26 38 ± 4 Genotype aEmbryos were scored at the 1.5-fold stage. The data shown are means s.e.m. Figures Figure 1. Wild-type and nuc-1 embryos show different TUNEL-staining patterns. (A) Wild-type embryo at the 1.5-fold stage (B) nuc-1(e1392) embryo at the same stage. Embryos were stained with TUNEL and photographed using a confocal microscope; each picture is a projection of eight serial confocal images. The white spots (some are indicated with white arrowheads) are TUNEL-positive nuclei. 164 Figure 1 Figure 2. nuc-1 cloning. (A) Genetic map location of nuc-1 (see Experimental Procedures). (B) A composite of the C. elegans nuc-1 transcript derived from genomic, cDNA and 5' RACE analysis. Intron positions are indicated by vertical bars. The deduced NUC-1 protein sequence is shown beneath. The arrow designates the base change in the three nuc-1 alleles. An inframe stop codon upstream of the predicted ATG and the termination codon are underlined. Asterisks indicate the locations of polyadenylation sites. (C) Deletion of C07B5.5 identified by PCR screening. Bracketed area indicates the extent of the deletion in n3335. (D) Alignment of NUC-1, C. elegans predicted proteins F09G8.2 and K04H4.6, and human DNase II (after Krieser and Eastman, 1998). Arrowhead indicates the site of cleavage in human DNase II. We have aligned the most similar nearby sequences in the worm proteins with the human DNase II cleavage site to indicate possible cleavage sites; it is not known whether the worm proteins are processed analogously. 166 Figure 2 IL 4 # to C~j c6 E co | | 1 m.u. B ACTCACTTCCCTTCTGAAACTTTCAAAATTTCAGCGGAAAAATGGGCTTGTCTCCTGCCGCTGTGCTGATTTTTCTCTTATTAGGAGTTTCCCAAACATATGCAGCATTCTCCTGCAAGGATCAGTCGGGAAACGATGTTGACTGETTCG M G L S P A A V L I F L L 392 Ae1 , n334, L G V S n887 Woo-stop Q T Y A A F S C K D Q G N D V D W F A CAGTGTACAAAATGCCAATCGAAAAAGATGATGGATCAGTGACAGGATTGGCAGGTGGAGTTGCATGGTACTACGTGGATGTGAATAAAAAGGGAACTTTGACTCCTTCTGCTAAAACTCTCGACGACAACGACCA*|CAATTGCATACA V Y K M P I E K D D G S V T G L A G G V A W Y Y V D V N K K G T L T P S A K T L D D N D Q A I A Y T CACTCCAACAATACTATGACAAGCAGAATGACAAGACCATCTTCCACGTGATGTACAATGATGA*CATGGGGATCAAAGTCGACAAGTGGAATTAAATTGGAAGAGATTCTGAGCAACAGAGTTTATAGCAACTATACTCATGAAGATG L Q Q Y Y D K Q N D K T I F H V M Y N D E P W G S K S T S G I K L E E I L S N R V Y S N Y T H E D ACAGTACTTCAACCGCATTCGGACATACTAAGGGAACCATTTTCTTTGATGGAACTTCTGGAGTCTGGTTGGTGCACAGTGTGCCATTGTTCCCAAATCCCACGAAATACGAGTATCCAGTCAGTGGACACGACTATGGACAGACAATGC S T S T A F G H T K G T I F D F G T S G V W L V H S V P L F P N P T K Y E Y P V S G H D Y G Q T M L TTTGCATGACATTTAAGTACGCTCAATTGAAGTCTATTGGAACTCAACTTTTCTTCAATCGACCAAATATATATTCTTCGAATCTTCCAACGAACATGGCTGCTGACAATGCAGATTTGGCTAAAGCAATCGCTGGACAATACCAAAAAG 150 37 300 87 450 137 600 187 G 750 237 GACAGCCATTCCAAAGCGTTATTGAGTTGGAGACTATGGCTGGATATTCGTTTACTAATTTTGCCAAGTCGAAGGAATTCAATGCA*TCTATACGATACTCTTGTGGCTCCAACACTGAAAACAGATTTGGTCGTTGAAACATGGAGAC 900 C Q M P T F F Q K S Y V A I Q E L L K E S T I M G A T G Q L Y S F F F T N N R F P A N K I S Y K S E S F N N L A P D T L N Y M D A T A L D V N A A P D T L L A K K T A D I L A V G V Q E Y T Q W K R R GTGGAAGTGAGATCCCACTTGACTGCAAACTAACCTACCACGCCAATGATGCTCTGTCAATTCA 4TTGGTAGTACAACCGCGTTCTCATACACCAAAGACCACTCGAAAATGGCTCACTCAGCTGATATGACAAAGCCGTGGGTGTGC G S E I P L D C K L T Y H A N D A L S I H V G S T T A F S Y T K D H S K M A H S A D M T K P W V C I TCGGAG.ATATCAACCGAATGNCATCTCAATATGTACGCGGAGGAGGTACTACTTGTATCAGCAGCTCGTTCCTTTGGAAAGCCTACAGTGTAATTGCAACCCAAAATAATTGTGCATAAAATTACATCTATGTACTTTTGATTATCTACA G D I N R M T S Q Y V R G G T G T C I S S S F L W AATAAATATGATTGATGTACTTCTT C n3335 r-- A Y S V I A T Q N N C A 1050 337 1200 375 1225 I I -- ON-- 250 bp nuc-1 K 287 Figure 2 D NUC-1 DNasell FO9G.2 OH4S6 NUC-1 DNasell KD4H4.6 1 1 1 1 MI4 L 22 25 II NM-----------------------------------------------------------------------------------------IFSVIFTTG ILESKICTQLVKTFSKNCANARQTLQKTPIKLGQMSHNSMIYLLVAFLVLLCATTEAKKECSIDCQEDGSAAVDLGLVLPPELYESTRLSNLLAR A3KIEVSET F IZE ------------------------------ ---------------------------------- --------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------D LIPSAKTL AW I - R SS-A---------------------K & K I GSKSTSGIKLEEILSNRVYSNYTHE - LAFL -------------------- LMPQG-MDVE NVQ H3SELAE !!IQAT NTFT NTV4A4AQY 4 ~ FQ N 4 l LTYANDALJSLVGSTTP I--- SD1IW LN ---------G EM ------------ l fl'.0E K~ IIj FPGUAG I EYN4DPNHVQSTSAKYIRLYAID PKG----- PL T --- 46 ELWNHPDNVPIS 240 rK- 172 "S 141 - 136 330 s GE §KV34LG3Q 4RH KT K F3 SSIK HQ 448 375 360 I3VEKSSSEFSMFQTIATLLSAAGLVSKI 347 516 SS] 0VIATNNCA LP 2P 45 288 259 250 wflf FTLMAUSLWTFEI PTl SLPRSAT FWII AAQ -LUSEAUS-- 47 -- t RI r--SSSKI; D11/G LA 54 ± SS KGHVSQ N--- SQLK NES - - S-- QP --------------- - tr tEI~D 173 142 137 331 289 260 251 449 119 C *QI MKS F9Ga2 KD4H42 ALLCVP 28 --- DQ------------------------------------------~----~ GD-------------------------------------------------~~---~------------------------------------- S 29 23 26 120 PSSQFKMKRDYDFVRFGRAAPIKKASYDYIRFAVPLFRKSIEGGFGCLDEIMFDIIGKGSTIQCQNIETTFHKMNLQNCLIALNFSLKKFQVSSDMQHFFL NUC-1 55 DNasell 47 FO9Ga2 48 K04H456 241 NUC-1 DNasell signal sequence MGLSPAAVLIFELGUSQTY LNY GMARKPSRAYKI Figure 3. n3335 TUNEL staining is like that of nuc-1(e1392). (A) Kinetics of TUNEL-reactive nuclei in nuc-1(e1392) embryos. (B) Kinetics of TUNELreactive nuclei in n3335 mutant embryos. The y axis indicates the number of TUNELpositive nuclei in stained embryos at the bean and comma (B/C), 1.5-fold (1.5-fold) or 3fold stage. Errors bars show standard deviations. 169 Figure 3 A 55- nuc- 1(e 1392) 5045- 40 U) C,. T I I 35- 3025 z 20|- 15- 6 10 z 5- 0 B/C B 55- 1.5-fold n3335 50- T 45- 40- 3-fold I T 350 CL zZ 302520- D 15- .. 6 10- z 5- 0B/C 1.5-fold 3-fold Figure 4. Unengulfed cell corpses in engulfment-defective mutants are TUNELpositive. (A) A ced-6(n1813); nuc-1(e1392) embryo stained with TUNEL. (B) The same embryo observed using Nomarski microscopy. A TUNEL-positive unengulfed cell corpse, which is excluded from the embryo, is indicated by an arrow. 171 Figure 4 Figure 5. Unengulfed cell corpses stain with SYTO 11. (A) A ced-6(n1813) L2 larva with unengulfed cell corpses (black arrowheads) in the ventral cord observed using Nomarski microscopy. (B) SYTO 11 staining of the same larva; staining is visible within live nuclei and those of the unengulfed corpses (white arrowheads). 173 Figure 5 Figure 6. Model for DNA degradation during programmed cell death in C. elegans. Each arrow represents a number of parallel or sequential nuclease activities that are mediated by the indicated genes. The final step likely requires the engulfment process per se, rather than a direct requirement for each of the engulfment genes in the DNA degradation process. Since mutations in ced-1 and ced-7 prevent engulfment, we propose that these genes act at the engulfment-mediated step in the pathway in addition to the initiation step (indicated by question marks). 175 Figure 6 ced-1 ced-7 Intact DNA > ced-2 ced-5 ced-6 ced-10 ced-1? ced-7? nuc-1 TUNEL-reactive DNA I TUN EL-un reactive DNA > Free nucleotides I I dying cell I engulfing cell Appendix 1 Genetics of programmed cell death in C. elegans: Past, present and future Mark M. Metzstein, Gillian M. Stanfield & H. Robert Horvitz 177 Abstract Genetic studies of the nematode Caenorhabditiselegans have defined a variety of single-gene mutations that have specific effects on programmed cell death. Analyses of the genes defined by these mutations have revealed that cell death is an active process that requires gene function in cells that die. Specific genes are required not only to cause cell death but also to protect cells from dying. Gene interaction studies have defined a genetic pathway for the execution phase of programmed cell death in C. elegans. Molecular and biochemical findings are consistent with the pathway proposed from these genetic studies and also have revealed that the protein products of certain celldeath genes interact directly. This pathway appears to be conserved among organisms as diverse as nematodes and humans. Important questions remain to be answered about C. elegans programmed cell death. For example, how does a cell decide to die? How is cell death initiated? What are the mechanisms of action of the cell-death protector and killer genes? What genes lie downstream of the cell-death execution pathway? The conservation of the central cell-death pathway suggests that additional genetic analyses of programmed cell death in C. elegans will help answer these questions not only for this nematode but also for other organisms, including ourselves. 178 Introduction The elimination of unwanted cells by programmed cell death is an important developmental and homeostatic process in multicellular organisms, including the nematode Caenorhabditiselegansl. During the development of the C. elegans hermaphrodite, 1090 cells are generated, of which 131 undergo programmed cell death 2 -4 (Fig. 1). Genetic analyses of C. elegans have identified genes that function in programmed cell death, have ordered these genes in a genetic pathway and have led to the finding that similar genes control programmed cell death in other organisms, including mammals. In this review, we concentrate on the genetic and developmental studies of C. elegans that have resulted in these discoveries. We mention only briefly molecular biological and biochemical studies of programmed cell death, which have been described in a number of recent reviews 5 -8 . We focus primarily on mechanisms of cell killing and for this reason do not discuss in detail the engulfment or degradation of cell corpses. egl-1, ced-4, ced-3 and ced-9 are global regulators of programmed cell death in C. elegans Three C. elegans genes, egl-1, ced-4 and ced-3, seem to be required for all somatic programmed cell death to occur. Loss-of-function (lf) mutations in egl-1, ced-4 or ced-3 lead to the survival of essentially all cells that undergo programmed cell death during wild-type development 9 ,1 0 . The egl-1 (egl, egg laying defective) gene was defined originally by gain-of-function (gf) mutations that cause a dominant egg-laying defect attributable to the loss of functional HSN neurons in hermaphroditesil, a defect found to be caused by the ectopic programmed deaths of these neurons 9 . The isolation of a cisdominant suppressor of the egl-1(gf) egg-laying defect revealed that the loss of egl-1 function causes an absence of programmed cell death1 0 . The first allele of ced-4 (ced, cell 179 death abnormal) was identified as an extragenic suppressor of the egg-laying defect of egl-1(gf) mutants 9 . ced-3 mutations were identified as suppressing the presence of persistent cell corpses seen in mutant animals defective in cell-corpse engulfment 9 . Subsequently, many more alleles of ced-4 and ced-3 have been obtained by screening for either the absence of cell corpses (e.g., Ref. 12) or the presence of extra living cells that would have died in wild-type animals (undead cells; e.g., Ref. 13) or by suppressing the lethality caused by loss-of-function mutations in the cell-death protection gene ced-9 (see below) (e.g., Ref. 14). The activity of the gene ced-9 protects most if not all cells from undergoing programmed cell death during C. elegans development. Loss-of-function mutations in ced-9 lead to sterility and maternal-effect lethality as a consequence of the ectopic activation of programmed cell death in cells that normally live 15 . In ced-9(lf) animals many cell types, including neurons, ectodermal cells and even undifferentiated blast cells, can activate the death program 15 . All of these deaths are suppressed by mutations in ced-4 and ced-3 (Ref. 15). ced-9 was defined originally by a gain-of-function allele that blocks programmed cell death (see below). Loss-of-function alleles were obtained by identifying cis-dominant suppressors. The molecular characterization of C. elegans cell-death genes has been key in revealing some of the biochemical mechanisms of programmed cell death in all animals. The discovery that ced-3 encodes a member of the caspase (cysteine aspartate-specific protease) family of proteases 16 ,1 7 was instrumental in identifying a role for mammalian caspases in the regulation of programmed cell death 5 . A protein related to the CED-4 protein, APAF-1, appears to be involved in effecting programmed cell death in mammalian cells 1 8 . Both the CED-4 and APAF-1 proteins may require nucleotide binding to function 1 9 -2 1 . The cloning of the ced-9 gene revealed that its protein product is similar to the human Bcl-2 protein 2 2 , which had been implicated in programmed cell death in mammalian cell culture experiments 2 3 . This discovery, along with the findings 180 that human Bcl-2 could block programmed cell death in C. elegans and could substitute for the C. elegans ced-9 gene in ced-9-deficient mutants 2 2 ,2 4 , strongly suggested that the molecular mechanism of programmed cell death is evolutionarily conserved. Finally, EGL-1 has functional and molecular similarity to the "BH3-only" (BH, Bcl-2 homology domain) subfamily of Bcl-2-like proteins: BH3-only proteins are capable of inducing programmed cell death when overexpressed in mammalian cell systems (Ref. 10 and references therein). egl-1 is the first member of the BH3-only family for which a loss-offunction phenotype has been described, indicating not only that a gene of this class can cause programmed cell death when overexpressed but also that such a gene normally functions in this way. Genetic evidence suggests that egl-1, ced-4 and ced-3 function exclusively to promote cell killing. Programmed cell death is not essential for the viability of C. elegans, and animals homozygous for loss-of-function mutations in egl-1, ced-4 or ced-3 appear superficially normal in size and movement 9 . Undead cells in C. elegans can differentiate 9 , 13 ,2 5 and can even function 2 6 . Loss-of-function alleles of ced-4 include some that are null by genetic and molecular criteria 1 2 . More than 90 alleles of ced-3 have been obtained of which more than 60 have been analyzed genetically and molecularly (Ref. 16 and H.R. Horvitz et al., unpublished results), and some are altered in residues essential for CED-3 enzymatic activity (Refs 16, 27 and S. Shaham, B. Davies, P. Reddien and H.R. Horvitz, unpublished results). However, no allele is known that is unambiguously predicted to eliminate expression of the entire CED-3 protein. Based on this consideration, it is conceivable that ced-3 has a role during the development of C. elegans unrelated to its enzymatic activity and that null alleles of ced-3 lead to additional (possibly lethal) defects. There is only one known loss-of-function allele of egl-1. This allele behaves as a null by genetic criteria, but molecular analysis indicates that at least a portion of the EGL-1 protein could still be produced, so it is not clear whether this mutation completely eliminates egl-1 activity 1 0 . 181 Since ced-9(lf) causes early lethality it is difficult to know whether ced-9 is required for any process other than the regulation of programmed cell death. However, double mutants between a null allele of ced-9 and either ced-3 or ced-4 do not appear to have any defects beyond those seen in ced-3 or ced-4 single mutants 1 4 , arguing that ced-9 function is as cell-death specific as are the functions of ced-3 and ced-4. Pathway to death Genetic experiments have ordered the functions of egl-1, ced-9, ced-4 and ced-3. First, as described above, loss-of-function mutations in ced-9 lead to lethality by causing the ectopic activation of programmed cell death. These deaths are suppressed by loss-offunction mutations in ced-4 or ced-3, indicating that ced-9 normally functions to negatively regulate ced-4 and ced-3 (Ref. 15). A simple pathway consistent with these observations places ced-9 genetically upstream of ced-4 and ced-3 (Fig. 2a). However, these observations do not exclude the possibility that ced-9 may function biochemically to inhibit the activities rather than the expression or activation of ced-4 and ced-3 (e.g., Ref. 28). By contrast, loss of egl-1 function, while preventing somatic cell death just as does loss of ced-4 or ced-3 function, does not suppress the lethality resulting from a loss of ced-9 function 1 0 . This finding suggests that egl-1 acts genetically upstream of and functions as a negative regulator of ced-9 (Fig. 2a). Since in a ced-9 loss-of-function mutant background the allelic states of ced-4 and ced-3 affect the fates of cells that would normally live, either the ced-4 and ced-3 gene products are normally present in most cells before the time at which cells differentiate or the loss of ced-9 activity leads to the ectopic expression of ced-4 and ced-3. Although the expression patterns of ced-4 and ced-3 at the RNA and protein levels have not been described, other molecular genetic evidence suggests that the former model is more likely to be correct. Specifically, transcriptional overexpression of ced-4 and/or ced-3 182 from transgenes in at least certain types of developing C. elegans neurons causes these cells to undergo programmed cell death 2 9 . The efficiency of this killing is dependent on the genotype of the animal carrying the transgene 2 9 . For instance, killing by ced-3 overexpression is more efficient when the transgenic animal is wild-type for ced-4 and/or mutant for loss-of-function of ced-9 (as described in more detail below). Since changes in the ced-4 or ced-9 genotype influence killing, ced-4 and ced-9 products are likely to be normally present in these cells 29 . Similar arguments suggest that ced-3 is also likely to be present. Transcriptional overexpression has also been used to order the functions of celldeath genes. Specifically, in the absence of ced-4 activity, ced-3 overexpression is able to kill cells, although at a reduced level compared to killing in a wild-type ced-4 background (Fig. 2b). By contrast, when ced-3 activity is reduced, ced-4 overexpression kills very inefficiently (Fig. 2b). These data suggest that ced-3 is needed for ced-4induced killing but that ced-4 is not needed for ced-3-induced killing. One simple model is that ced-4 potentiates ced-3 activity and that a sufficiently high level of ced-3 can bypass the normal requirement for ced-4 activity. Similar results have been obtained by the overexpression of ced-4 and ced-3 in insect cells 3 0 . Why then is there any killing by overexpressed ced-4 in a ced-3(lf) background? One possibility is that killing is a consequence of ced-4-induced activation of the residual activity of the ced-3 alleles tested (as described above, none of the known ced-3 alleles is definitively a null mutation). Another possibility is that the overexpression of ced-4 leads to the apparent absence of the cell not because the cell has died but rather because it is misplaced or not generated. We favor a third possibility: perhaps overexpressed ced-4 can trigger programmed cell death independently of ced-3, either through another caspase or independently of caspase activity. At least two additional genes with similarity to ced-3 are present in C. elegans genomic sequences (P. Reddien and H.R. Horvitz, unpublished results; M. Hengartner, personal communication). 183 Biochemical data have supported the hypothesis that ced-4 is an upstream activator of ced-3 and have provided a possible physical basis for their interaction. The CED-4 protein may directly bind to the pro-enzyme form of the CED-3 protein to facilitate its proteolytic conversion to the active enzyme 20,31,32. In mammals APAF-1 (a CED-4related protein) binds to and stimulates the processing of caspase-9 (a CED-3-like protein) 1 8 ,2 1 . High levels of CED-3 may be able to bypass the need for CED-4 to facilitate conversion from the pro-enzyme form, explaining how ced-3 overexpression can kill in the absence of ced-4 activity. Similar overexpression experiments suggest that ced-9 acts by inhibiting ced-4 function 2 9 (Fig. 2c). The presence of an endogenous wild-type ced-9 allele partially inhibits the ability of overexpressed ced-3 to kill cells that normally live: killing by ectopically expressed ced-3 is less efficient in a ced-9 wild-type animal than in a ced-9 loss-of-function animal. However, in a ced-4 mutant background, killing by ced-3 is no less efficient in animals carrying a wild-type ced-9 allele than in animals carrying a ced-9(lf) allele. These results indicate that in the absence of ced-4 activity, ced-9 fails to protect against killing by overexpressed ced-3. Thus, ced-9 protection against ced-3 killing is mediated at least in part by ced-4 (Fig. 2c). Results consistent with this hypothesis were also obtained by examining the effects of expression of combinations of ced-9, ced-4 and ced-3 in insect and mammalian cells 3 0 ,3 1 . A physical basis for the results of these genetic studies is suggested by the observation that CED-9 and CED-4 can interact directly (reviewed in Ref. 33). This interaction has been proposed to block the activity of CED-4, although the mechanism of such an inhibition is unknown. An interaction between CED-9 and CED-4 has been demonstrated in vitro and in yeast, insect and mammalian cells but not yet directly in C. elegans, although some ced-9 and ced-4 allele-specific interactions have been observed 34. The human CED-4-like protein APAF-1 and a Bcl-2 family member, Bcl-xL, may also interact directly 3 5 ,3 6 . 184 Overexpression of egl-1 can kill cells that normally live 10 . This killing is dependent on ced-4 and ced-3, placing egl-1 genetically upstream of these genes (Fig. 2d). As described above, egl-1 also seems to function upstream of and as a negative regulator of ced-9. The EGL-1 protein binds to the CED-9 protein, and it has been proposed that EGL-1 acts by releasing CED-4 from CED-9 (Ref. 10). Similarly, in mammals BH3-only proteins bind Bcl-2 family proteins (various references cited in 10). Thus, current genetic and molecular data suggest the following model for the activation of programmed cell death (Fig. 3). After activation by upstream signals, the EGL-1 protein interacts with CED-9 and releases CED-4 protein from membraneassociated CED-9 protein (CED-9, like other Bcl-2 family members, has a hydrophobic C-terminus that likely causes it to be membrane-associated 3 7 .) Free CED-4 then interacts with and facilitates the processing of inactive pro-CED-3 to the active enzyme. The active caspase acts as the mediator of downstream events in cell death, eventually leading to the destruction of the cell by cleaving and thereby activating additional killing proteins and/or by cleaving and thereby inactivating additional protecting proteins or proteins needed for cellular homeostasis. ced-9 and ced-4 have both protecting and killing activities Genetic evidence suggests that in addition to its protective role ced-9 also may activate programmed cell death, at least in cells that normally die. Specifically, the survival of cells in weak ced-3 mutants is enhanced by loss-of-function alleles of ced-9 (Ref. 14). Two genes related to ced-9, Bcl-x and Bcl-2, each encode opposing cell-death activities, but there is no evidence that the opposing activities of ced-9 are generated either by alternative splicing, as for Bcl-x (Ref. 38), or by proteolytic processing, as for Bcl-2 (Ref. 39). A gain-of-function allele of ced-9, n1950, prevents programmed cell death, just as do egl-1, ced-4 and ced-3 loss-of-function mutations1 5 . Overexpression of 185 wild-type ced-9 can also block programmed cell death 1 4 ,2 2 ,2 8 , so it is possible that ced-9(n1950) acts to increase an essentially wild-type ced-9 activity. Alternatively, it is possible that the n1950 mutation inactivates the killing function of ced-9 while leaving its protective function intact. Since the ced-9(n1950) mutation alters the BH1 domain of the CED-9 protein, a domain that is known to be involved in protein-protein interactions in other CED-9/Bcl-2-family proteins4 0, 4 1 , one possibility is that the n1950 mutation alters the specificity of protein-protein interactions. It should be noted, however, that the equivalent change in the CED-9 homologue Bcl-2 behaves as a loss-offunction mutation and fails to protect against cell death both in mammalian cells 41 and in C. elegans14 . Thus, one should be cautious about drawing conclusions concerning the function of the CED-9 BH1 domain based upon other Bcl-2 family members and, more generally, concerning the function of the BH1 domain of any Bcl-2 family protein based upon any other family member. ced-4 also can function both as a cell-death activator and as a cell-death inhibitor. These opposing activities appear to result from two protein isoforms of CED-4 encoded by alternatively spliced transcripts 3 4 . The discovery of an alternatively spliced ced-4 transcript was based upon comparative genomic studies. Introns in C. elegans and the related nematode species Caenorhabditisbriggsae or Caenorhabditisremanei (formerly C. vulgaris4 2 ) tend to be poorly conserved in sequence (although not in position) 4 2 . The ced-4 gene appeared to be an exception, since within the third intron there is a 72nucleotide region of very high (>90%) identity among all three species. This region proved to correspond to an alternatively spliced exon that is present in a second, less abundant, ced-4 transcript and that is predicted to produce a protein, CED-4L (L, long), with 24 additional amino acids inserted between residues 212 and 213 of the 549 amino acid killing form, CED-4S (S, short). Whereas when overexpressed from a transgene CED-4S causes programmed cell death, when overexpressed from a transgene CED-4L can protect against programmed cell death. ced-4L overexpression can also protect 186 against ectopic programmed cell deaths caused by ced-9 loss of function, indicating that ced-9 functions genetically upstream of ced-4L, just as it functions genetically upstream of ced-4S. It is not known how the splicing choice is controlled or whether this control is differentially regulated in different cells (e.g., those that normally live and those that normally die) to alter their susceptibility to death-inducing signals or perhaps to initiate the cell death program. Furthermore, how the CED-4L protein inhibits programmed cell death is unknown. The simplest model is that CED-4L functions as an interfering dominant-negative form of the CED-4 protein: for example, CED-4L could interact with the same targets as does CED-4S but not activate these targets, while blocking CED-4S from doing so. Consistent with this hypothesis, recent biochemical evidence suggests that CED-4L can bind to but not promote the processing of the pro-enzyme form of CED-3 (Ref. 43). It is possible that the killing function of ced-9 and the protective function of ced-4 are related. CED-9 may kill by binding to and inhibiting the activity of CED-4L, much in the same way that CED-9 protects by binding to and inhibiting the activity of CED-4S (see Ref. 34). Developmental regulation of programmed cell death How does a cell decide to undergo programmed cell death? In mammals, cell interactions act to trigger at least some programmed cell deaths. In C. elegans, a few cell deaths depend upon interacting cells 4 4 , but many seem likely to be cell-autonomous 4 5 . How might such cell-autonomous deaths be initiated? We suggest that there are two ways to think about the existing observations. First, programmed cell deaths might be triggered by an underlying cellular defect, such as a defect in differentiation. Second, programmed cell death might be a differentiated fate expressed as a consequence of the normal process of selecting among a set of differentiation choices. Undergoing 187 programmed cell death is similar in a number of ways to adopting any differentiated fate, such as becoming a neuron or muscle cell. Like other cell fates in C. elegans, cell deaths occur throughout the cell lineage (Fig. 1), and the majority of cell deaths occur during the developmental period when most other cells terminally differentiate 2 ,4 . Also, like other cell fates in C. elegans, programmed cell death is observed in an essentially invariant pattern with the same lineally equivalent cells undergoing programmed cell death from animal to animal. This reproducibility in the cells that die suggests that programmed cell death in C. elegans is not a result of stochastically occurring defects in cellular physiology. Furthermore, patterns of programmed cell deaths can be altered by mutations in genes known to be involved in the control of other cell fates (e.g., Refs 46-48), further indicating that programmed cell death is no different from developmental cell fates in general. How do developmental signals control the activities of the killing machinery, which includes at least egl-1, ced-9, ced-4 and ced-3, to initiate the death process in particular cells? Although some understanding has been reached of how certain cell-cell interactions in the mammalian immune system lead to the activation of the cell-death machinery (reviewed in Ref. 49), much less is known about how the machinery is activated during development in C. elegans or in any other organism. One possibility is that the activity of one or more of these genes is under transcriptional control. Although many living cells in C. elegans probably express ced-4 and ced-3, transcriptional overexpression of either of these genes, or of egl-1, is sufficient to cause the deaths of at least some cell types1 O, 2 9 . Thus, elevation of egl-1, ced-4 and/or ced-3 transcription could be a mechanism used to initiate programmed cell death. The direct transcriptional regulation of the ced-9 gene does not seem as likely to be responsible for initiating cell death (at least during embryogenesis), since this gene shows a strong maternal component and no zygotic ced-9 activity is required for the normal regulation of programmed cell death during embryogenesis 15 . 188 Genetic analysis has identified two genes, ces-1 and ces-2 (ces, cell death specification) that control a subset of programmed cell deaths in C. elegans. While mutations in ces-1 or ces-2 block the deaths of certain neural cells 13 , no other discernible cell-death, cell-lineage or cell-fate defects have been observed in ces-1 or ces-2 mutants, suggesting that ces-1 and ces-2 are specifically involved in regulating the programmed cell deaths of these cells. ces-2 encodes a member of the basic-leucine zipper (bZIP) family of transcription factors 5 0 , consistent with the hypothesis that programmed cell death is controlled at the level of differential gene expression. Mammalian members of the bZIP family most similar to CES-2 (the proline and acid rich or PAR family) also may have a role in the cell-specific regulation of programmed cell death 50 ,5 1 . It is conceivable that CES-2 is a direct transcriptional regulator of egl-1, ced-4 or ced-3. It is important to note, however, that the Ces phenotype of ces-1 mutant animals is caused by gain-of-function alleles and that of ces-2 animals is caused by a partial loss-offunction allele 1 3 . Since these alleles do not result in a complete loss of gene activity, it remains possible that the cell-specific cell-death phenotypes of ces-1 and ces-2 animals represent an aspect of a more general requirement for these genes in programmed cell death or in cell-fate determination. Finally, the regulation of ced-9 activity is unlikely to be the only mechanism controlling the life or death decision. Animals doubly mutant for a null allele of ced-9 and weak alleles of ced-3 are viable and do not have a large number of ectopic cell deaths 1 4 . However, in such animals the majority of cells that normally die during wildtype development still do so14, indicating that in this genetic background cells that normally die are more susceptible to programmed cell death than are cells that normally live. Since ced-9 is inactive in these mutants, this difference in susceptibility cannot arise from differential regulation of ced-9 activity. Hence, some death-promoting signals must feed into components of the cell-death pathway independently of ced-9. 189 In the throes of death As cells undergo programmed cell death, their corpses are rapidly engulfed by and then degraded within engulfing cells. In C. elegans most corpses are engulfed by their closest neighbors (Ref. 52 and J. Sulston and J. White, personal communication). Six genes, ced-1,-2,-5,-6,-7 and -10, important for cell-corpse engulfment in C. elegans, have been discovered in genetic screens by visually identifying mutants that contain unengulfed cell corpses 5 3 ,5 4 . (The gene ced-8, which was originally classified as an engulfment ced gene, instead is involved in a different aspect of the cell-death process; G.M. Stanfield, M. Hengartner and H.R. Horvitz, unpublished observations.) Recent papers describe molecular analyses of three of these engulfment genes 5 5 -5 7 . At least one gene, nuc-1 (nuc, nuclease), is involved in the degradation of the DNA of cell corpses 5 8 . In nuc-1 animals, the condensed DNA of cell corpses persists indefinitely within engulfing cells. nuc-1 appears to encode or control the activity of a nuclease that acts not only in cell death but also in other processes, since nuc-1 mutants are unable to digest bacterial DNA in their guts and have a large reduction in at least one biochemically-defined endonuclease activity present in wild-type animals 5 3 ,5 9 . Mutations in nuc-1 or in any of the engulfment genes, alone or in combination, do not affect the execution of cell deaths. The wild-type pattern of cell deaths occurs, and the timing of the appearance of cell corpses is not altered 5 3 . Thus, the activities of these genes are not required for cell killing. One model consistent with these data is that multiple, independent activities are required to effect different aspects of the postexecution death program, such as the morphological changes in the dying cell, engulfment and cellular degradation. Loss of any one of these functions is not sufficient to block programmed cell death in C. elegans. It is also possible that mutations in nuc-1 and/or in the engulfment ced genes affect programmed cell death nonspecifically; that 190 is, these genes are involved in general cellular functions in both living and dying cells and dying cells are more sensitive than are living cells to defects in those functions. Beyond the Valley of the Shadow of Death Genetic analysis has led to the identification of key central regulators of programmed cell death in C. elegans, and molecular and biochemical studies are providing clues about the mechanisms of action of these genes and their protein products. Nonetheless, many aspects of the regulation and function of the egl-1, ced-9, ced-4 and ced-3 genes are still unknown. For instance, ces genes have been identified for only a small subset of cell types. These cell types may have unique cell-death controls, but it seems more likely that many ces mutants have been overlooked because their phenotypes are subtle. How is egl-1, the most upstream component of the general cell death machinery, regulated? How does CED-4 trigger the conversion of inactive CED-3 to the active form? How are relative levels of CED-4L and CED-4S controlled? What is the basis for the killing activity of ced-9 and the protective activity of ced-4L? What are the steps downstream of CED-3 activation that mediate cell-corpse formation, engulfment and degradation? Further genetic analysis using C. elegans should help reveal the answers to these questions. While there have been extensive screens to identify mutations that can suppress the lethality conferred by ced-9(lf) mutations (M. Hengartner, S. Shaham, B. Davies, L. Speliotes, A. Madi and H.R. Horvitz., unpublished results), there has been only limited screening for genes that, like egl-1, function to promote cell death upstream of ced-9. Furthermore, most screens for cell-death mutants have required that animals be viable, so other protector genes like ced-9 might not have been identified. Genes with redundant functions in cell killing also would likely have been missed in previous screens. Searching for enhancers or suppressors of cell killing in genetically sensitized 191 backgrounds (e.g., Ref. 60) or examining cell death in animals carrying chromosomal deletions (e.g., Refs 61,62) offer two approaches to the identification of such genes. What are the targets of CED-3 proteolysis? There may be a small number of critical targets that are activated to effect the downstream events of cell death. Alternatively, there may be multiple targets with important cellular functions that are inactivated by CED-3 proteolysis. This latter model assumes that although cell death initiates as a regulated process, it proceeds by eliminating functions required for normal cellular homeostasis. Another possibility is that ced-3 functions to inactivate a cell-death protector. Loss of function of such a gene would presumably confer a lethal phenotype and would not be suppressed by any known cell-death mutant. The identification and characterization of genes involved in the generation of the conserved morphological changes in cell corpses, in cell-corpse degradation and in the generation of engulfment signals might reveal which, if any, of these models is true. Few genetic screens have sought genes involved in corpse formation and breakdown. Perhaps targets of the execution genes could be sought by screening for mutations that synthetically alter cell death patterns, for example by blocking cell death when in combination with mutations that cause other downstream defects. While numerous proteolytic targets of mammalian caspases have been identified 6 3 , the in vivo relevance of these targets to programmed cell death has been mostly untested. The genetic analysis of such genes in C. elegans should allow their functions to be determined. Conclusions Enormous progress has been made toward understanding the basic molecular mechanisms used by cells to kill themselves. Nonetheless, many questions remain. The remarkable degree of conservation of the cell-death pathway from nematodes to mammals suggests that genetic analysis of cell death in C. elegans will continue to play a 192 major role in revealing the mechanisms responsible for this crucial and fascinating process. 193 Acknowledgements We wish to thank T. Herman, J. Agapite and myriad members of the Horvitz laboratory for helpful comments concerning the manuscript. H.R.H. is an Investigator of the Howard Hughes Medical Institute. 194 References 1 Ellis, R.E., Yuan, J. and Horvitz, H.R. (1991) Annu. Rev. Cell Biol. 7, 663-698 2 Sulston, J.E. and Horvitz, H.R. (1977) Dev. Biol. 56, 110-156 3 Kimble, J. and Hirsh, D. (1979) Dev. Biol. 70, 396-417 4 Sulston, J.E., Schierenberg, E., White, J.G. and Thomson, J.N. (1983) Dev. Biol. 100, 64-119 5 Thornberry, N.A. (1997) Br. Med. Bull. 53,478-490 6 Kidd, V.J. (1998) Annu. Rev. Physiol. 60, 533-573 7 Newton, K. and Strasser, A. (1998) Curr. Opin. Genet. Dev. 8, 68-75 8 Bergeron, L. and Yuan, J. (1998) Curr. Opin. Neurobiol. 8, 55-63 9 Ellis, H.M. and Horvitz, H.R. (1986) Cell 44, 817-829 10 Conradt, B. and Horvitz, H.R. (1998) Cell 93, 519-529 11 Trent, C., Tsung, N. and Horvitz, H.R. (1983) Genetics 104, 619-647 12 Yuan, J. and Horvitz, H.R. (1992) Development 116, 309-320 13 Ellis, R.E. and Horvitz, H.R. (1991) Development 112, 591-603 14 Hengartner, M.O. and Horvitz, H.R. (1994) Nature 369, 318-320 15 Hengartner, M.O., Ellis, R.E. and Horvitz, H.R. (1992) Nature 356, 494-499 16 Yuan, J. et al. (1993) Cell 75, 641-652 17 Alnemri, E. et al. (1996) Cell 87, 171 18 Zhou, H. et al. (1997) Cell 90, 405-413 19 James, C., Gschmeissner, S., Fraser, A. and Evan, G.I. (1997) CurrBiol 7, 246-52 20 Chinnaiyan, A.M. et al. (1997) Nature 388, 728-729 21 Li, P. et al. (1997) Cell 91, 479-489 22 Hengartner, M.O. and Horvitz, H.R. (1994) Cell 76, 665-676 23 Vaux, D.L., Cory, S. and Adams, J.M. (1988) Nature 335,440-442 24 Vaux, D.L., Weissman, I.L. and Kim, S.K. (1992) Science 258, 1955-1957 195 25 White, J.G., Southgate, E. and Thomson, J.N. (1991) Philos. Trans. R. Soc. Lond. Ser B 331, 263-271 26 Avery, L. and Horvitz, H.R. (1987) Cell 51, 1071-1078 27 Xue, D., Shaham, S. and Horvitz, H.R. (1996) Genes Dev. 10, 1073-1083 28 Xue, D. and Horvitz, H. (1997) Nature 390, 305-308 29 Shaham, S. and Horvitz, H.R. (1996) Genes Dev. 10, 578-591 30 Seshagiri, S. and Miller, L.K. (1997) Curr. Biol. 7,455-460 31 Wu, D., Wallen, H.D., Inohara, N. and Nunez, G. (1997) J. Biol. Chem. 272, 21449- 21454 32 Chinnaiyan, A.M., O'Rourke, K., Lane, B.R. and Dixit, V.M. (1997) Science 275, 1122-1126 33 Vaux, D.L. (1997) Cell 90, 389-390 34 Shaham, S. and Horvitz, H.R. (1996) Cell 86, 201-208 35 Pan, G., O'Rourke, K. and Dixit, V.M. (1998) J. Biol. Chem. 273, 5841-5845 36 Hu, Y. et al. (1998) Proc. Natl. Acad. Sci. U. S. A. 95, 4386-4391 37 Nguyen, M. et al. (1993) 38 Boise, L.H. et al. (1993) Cell 74,597-608 39 Cheng, E.H. et al. (1997) Science 278, 1966-1968 40 Muchmore, S.W. et al. (1996) Nature 381, 335-341 41 Yin, X.-M., Oltvai, Z.N. and Korsmeyer, S.J. (1994) Nature 369, 321-323 42 Riddle, R.L., Blumenthal, T., Meyer, B.J. and Priess, J.R., eds (1997) C. elegans II 43 Chaudhary, D., O'Rourke, K., Chinnaiyan, A.M. and Dixit, V.M. (1998) J Biol J. Biol. Chem. 268, 25265-25268 Chem 273, 17708-12 44 Sulston, J.E., Albertson, D.G. and Thomson, J.N. (1980) Dev. Biol. 78, 542-576 45 Sulston, J.E. and White, J.G. (1980) Dev. Biol. 78,577-597 46 Wang, B.B. et al. (1993) Cell 74, 29-42 47 Clark, S.G., Chisholm, A.D. and Horvitz, H.R. (1993) Cell 74, 43-55 196 48 Guenther, C. and Garriga, G. (1996) Development 122, 3509-3518 49 Cohen, G.M. (1997) Biochem. J. 326, 1-16 50 Metzstein, M.M. et al. (1996) Nature 382, 545-547 51 Inaba, T. et al. (1996) Nature 382,541-544 52 Robertson, A.M.G. and Thomson, J.N. (1982) J. Embryol. Exp. Morphol. 67, 89-100 53 Hedgecock, E., Sulston, 54 Ellis, R.E., Jacobson, D.M. and Horvitz, H.R. (1991) Genetics 129, 79-94 55 Liu, Q.A. and Hengartner, M.O. (1998) Cell 93, 961-72 56 Wu, Y.C. and Horvitz, H.R. (1998) Nature 392, 501-504 57 Wu, Y.C. and Horvitz, H.R. (1998) Cell 93, 951-60 58 Sulston, J.E. (1976) Philos. Trans. R. Soc. London Ser. B 275, 287-298 59 Hevelone, H. and Hartman, P.S. (1988) Biochem. Genet. 26,447-461 60 Hay, B.A., Wassarman, D.A. and Rubin, G.M. (1995) Cell 83, 1253-1262 61 White, K. et al. (1994) Science 264, 677-683 62 Terns, R.M. et al. (1997) Genetics 146, 185-206 63 Rosen, A. and Casciola-Rosen, L. (1997) 64 Wood, W.B. and The Community of C. elegans Researchers. (1988) The Nematode J. and Thomson, J.N. (1983) Science 220, 1277-1279 J. Cell. Biochem. 64, 50-54 Caenorhabditiselegans 65 Nicholson, D.W. and Thornberry, N.A. (1997) Trends Biochem. Sci. 22, 299-306 197 Figures Figure 1. Distribution of programmed cell deaths within the cell lineage of the C. elegans hermaphrodite. Programmed deaths occur in most of the major developmental lineages (only the E cell lineage, which produces only intestine, and the D cell lineage, which produces only muscle, lack programmed cell deaths) 2 ,4,4 4 . Cell death is a particularly common fate in the AB cell lineage (116 of the 722 cells generated proceed to die), which also produces most of the nervous system. In this figure, horizontal lines indicate early embryonic cell divisions, colors represent cell fates and areas are proportional to the total numbers of embryonic and postembryonic descendents from the primary blastomeres AB, MS, E, C and D. The germline, generated from blast cell P4, unlike other tissues in C. elegans, continuously proliferates and gives rise to variable numbers of cells and cell deaths 3 ,6 4 . Adapted from Ref. 4. 198 Figure 1 * Nervous system and other ectoderm * Muscle and other mesoderm * Intestine 0 Programmed Cell Deaths Zygote AB MS E C D ih #A 40 P4 No. surviving cells: 606 252 34 47 20 Variable No. programmed cell deaths: 116 14 0 1 0 Variable Figure 2. Interactions among egl-1, ced-9, ced-4 and ced-3. Genetic epistasis experiments and studies in which egl-1, ced-9, ced-4 and ced-3 have been overexpressed to kill cells in different genetic backgrounds have suggested an order of action of these genes1 0 ,15 ,29 . The relative efficiency of cell killing is represented by the number of plus (+) signs, as interpreted from data in Refs 10, 29. a) Loss-of-function mutations in ced-4 or ced-3 suppress the lethality caused by loss of ced-9 function, suggesting that ced-9 functions upstream of ced-4 and ced-3. By contrast, a loss of function mutation in egl-1 does not suppress ced-9(lf) lethality, suggesting that egl-1 functions upstream of ced-9. b) The overexpression of ced-3 or ced-4 can kill cells in wild-type C. elegans. In the absence of ced-4 activity, overexpressed ced-3 can still kill, albeit at a slightly reduced efficiency. By contrast, in the absence of ced-3 activity, ced-4 is almost incapable of killing cells. These results suggest ced-4 acts upstream of ced-3 to activate programmed cell death. c) Endogenous ced-9 activity protects cells from programmed cell death induced by overexpression of ced-3, since killing is more efficient in a ced-9(lf) background than a ced-9(+) background. While elimination of ced-4 function reduces the killing activity of ced-3, this residual ced-3 activity is no longer inhibited by ced-9, suggesting that ced-9 functions to protect against programmed cell death at least in part by negatively regulating ced-4. d) The overexpression of egl-1 can also kill cells. This killing is blocked by loss-offunction mutations in ced-4 or ced-3 or by a gain-of-function mutation in ced-9, suggesting that egl-1 acts upstream of these genes. 200 Figure 2 a) Genotype Phenotype Viable? Programmed cell death Wild-type Yes Normal ced-9(lf) No Excess ced-4(lf) Yes Reduced ced-3(lf) Yes Reduced egl-1(lf) Yes Reduced ced-4(lf) ced-9(lf) Yes Reduced ced-9(lf); ced-3(lf) Yes Reduced ced-9(lf); egl-1 (lf) No Excess egl-1 - - ced-9 -- ced-4 ce d-3 Programmed cell death b) Overexpressed gene Genotype ced-3 ced-4(+) ced-4 ced-3(+) ced-3 ced-4(lf) ++ ced-4 ced-3(lf) +1- ced-4 -- ced-3 o- Cell killing Programmed cell death c) Genotype ced-4 ced-9 ced-3-induced killing + if ++++ ced-9 protection + if if if + ++ ced-9 -- d) ced-4 - Genotype N No ced-9 protection ced-3 egl-1-induced killing Wild-type ced-3(lf) ced-4(lf) ced-9(gf) egl-1 - ced-9 -- ced-4 -- + ced-3 Figure 3. A model for the activation of programmed cell death in C. elegans. Step 1. Upstream signals lead to the production or activation of the EGL-1 protein. EGL-1 binds to CED-9, shown localized to cell membranes, leading to the release of CED-4 from CED-9. Although shown as a monomer, CED-9 likely functions as a dimer, as do other Bcl-2 family proteins 7 . Step 2. Free CED-4 then promotes the proteolytic cleavage of the pro-enzyme form of CED-3; p17 and p13 subunits derived from this processing assemble into the active form 2 7 (active caspases are thought to be tetramers, consisting of dimers of such a heterodimer; reviewed in Ref. 65.) It is possible that CED-3 is complexed with CED-4 before EGL-1 mediates the release of CED-4 from CED-9. 203 Figure 3 Step 2: Step 1: , Upstream signals EGL-1 GL-1 1' 2ECleavage Inactive CED-3 Active CED-3