In symptomatic generalized epilepsies, absence seizures are due to
advertisement

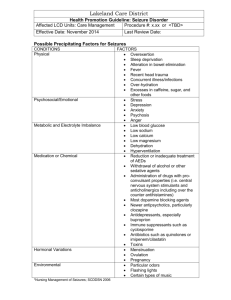
ABSENCE SEIZURES Introduction Background Absence seizures are a type of generalized seizures.1,2 They were first described by Poupart in 1705, and later by Tissot in 1770, who used the term petit access. In 1824, Calmeil used the termabsence.3 In 1935, Gibbs, Davis, and Lennox described the association of impaired consciousness and 3-Hz spike-and-slow-wave complexes on electroencephalograms (EEGs).4 Absence seizures occur in both idiopathic and symptomatic generalized epilepsies.5 Among the idiopathic generalized epilepsies, absence seizures are seen in childhood absence epilepsy (pyknolepsy), juvenile absence epilepsy, and juvenile myoclonic epilepsy (impulsive petit mal).6The seizures in these conditions are called typical absence seizures and are usually associated with generalized 3-4 Hz spike-and-slow-wave complexes on EEG.7 In childhood absence epilepsy, seizures are frequent and brief, lasting just a few seconds (pyknoleptic). Some children can have many such seizures per day. In other epilepsies, particularly those with an older age of onset, the seizures can last several seconds to minutes and may occur only a few times a day (called nonpyknoleptic or spanioleptic absence seizures). Myoclonic and tonic-clonic seizures may also be present, especially in syndromes with an older age of onset. In these syndromes, the discharge frequency may be faster than 3 Hz. In the cryptogenic or symptomatic generalized epilepsies, absence seizures are often associated with slow spike-wave complexes of 1.5-2.5 Hz6 ; these are also called sharp-and-slowwave complexes. These seizures may be associated with loss of axial tone and head nodding or a fall may occur. Increased tone, autonomic features, and automatisms may also be seen. Absence seizures associated with slow spike-wave complexes are called atypical absence seizures.8 Pathophysiology Etiology The etiology of idiopathic epilepsies with age-related onset is genetic. About 15-40% of patients with these epilepsies have a family history of epilepsy; overall concordance in monozygotic twins is 74% with a 100% concordance during the peak age of phenotypic expression.9 Family members may have other forms of idiopathic or genetic epilepsy (eg, febrile convulsions, generalized tonic-clonic seizures). Several animal models demonstrate the genetic basis for absence seizures. A strain of Wistar rat, genetic absence epilepsy rats from Strasbourg (GAERS), is a polygenetic model10 in which all animals have clinical seizures consisting of a behavioral arrest with twitching of facial muscles. This is associated with bilateral synchronous spike-wave discharges. Several single-gene loci in mice, when mutated, result in generalized spike-wave epilepsy. The tottering (chromosome 8), lethargic (chromosome 2), stargazer (chromosome 15), mocha (chromosome 10), and ducky (chromosome 9) loci all have generalized 6-per-second spike-wave EEG paroxysms that are associated with clinical seizures consisting of behavioral arrest. All types respond to ethosuximide, but the underlying cellular mechanisms for the generation of the discharges are not identical.11 The idiopathic generalized epilepsies are a group of primary generalized epilepsies with absence, myoclonic, and tonic-clonic seizures. Based on age of onset and seizure types, some can be grouped into well-recognized syndromes such as childhood absence epilepsy, juvenile absence epilepsy, and juvenile myoclonic epilepsy, but other syndromes such as generalized epilepsy with febrile seizures plus (GEFS+), or patients who have childhood absence epilepsy that leads into juvenile myoclonic epilepsy illustrate that these syndromes represent a genetically determined lower threshold to have seizures. The idiopathic generalized epilepsies are best viewed as a spectrum of clinical syndromes12 with varied genetic causes that affecting the function of ion channels. Genetic studies have shown that these syndromes are channelopathies, but different gene mutations have been found in the same syndromes. Juvenile myoclonic epilepsy has been linked to chromosome 613,14 with linkage to chromosome 6p12 in Mexican families15 . More recently, mutations in the EFHC1 gene were found in Mexican16,17 and Italian families18 with juvenile myoclonic epilepsy, but not in a group of Dutch families19 . Childhood absence epilepsy with generalized tonic-clonic seizure has been linked to chromosome 8q24 in a 5-generation family from Bombay, India.20 Childhood absence epilepsy with febrile seizures has been linked to the GABA(A) receptor γ2 subunit (GABRG2) on chromosome 5q3.1-33.121 . More recently, a mutation in the GABA(A) receptor gene GABRB3 was found in Mexican families with childhood absence epilepsy. Mutations showed hyperglycosylation in vitro with reduced GABA-evoked current density from whole cells. Expression of this gene in the developing brain may help explain an age-related onset and remission in childhood absence epilepsy.22 In symptomatic generalized epilepsies, absence seizures are due to a wide variety of causes that, at an early stage of neural development, result in diffuse or multifocal brain damage. The causes of secondary generalized epilepsies and the other seizure types that accompany them, and their management are discussed elsewhere (Epilepsy in Children with Mental Retardation, Lennox-Gastaut Syndrome), and are not discussed in this article. Pathophysiology The pathophysiology of absence seizures is not fully understood. In 1947, Jasper and Droogleever-Fortuyn electrically stimulated nuclei in the thalami of cats at 3 Hz and produced bilaterally synchronous spike-and-wave discharges on EEG.23 In 1953, bilaterally synchronous spike-and-wave discharges were recorded by using depth electrodes placed in the thalamus of a child with absence seizures.24 In 1977, Gloor demonstrated that the bilaterally synchronous 3-Hz spike-wave discharges in the feline penicillin model of absence seizures were generated in the cortex.25 This led to the corticoreticular theory of primarily generalized seizures. Abnormal oscillatory rhythms are believed to develop in thalamocortical pathways. This involves GABA-B–mediated inhibition alternating with glutamate-mediated excitation. The cellular mechanism is believed to involve T-type calcium currents. T channels of the GABAergic reticular thalamic nucleus neurons appear to play a major role in the spike-wave discharges of the GABAergic thalamic neurons.26 GABA-B inhibition appears to be altered in absence seizures, and potentiation of GABA-B inhibition with tiagabine (Gabitril), vigabatrin (Sabril), and possibly gabapentin (Neurontin) results in exacerbation of absence seizures. Enhanced burst firing in selected corticothalamic networks may increase GABA-B receptor activation in the thalamus, leading to generalized spike-wave activity. Frequency United States The incidence is 1.9-8 cases per 100,000 population. Mortality/Morbidity No deaths result directly from absence seizures. Accidents from driving or operating dangerous machinery during absence may result in death. In children with absence seizures due to secondary generalized epilepsies, death is related to the underlying disease. The morbidity from typical absence seizures is related to the frequency and duration of the seizures, as well as to the patient's activities; effective treatment ameliorates these factors. Educational problems and behavioral problems are sequelae of unrecognized, frequent seizures. Race No racial predilection is known. Sex Absence seizures are generally believed to be more common in females and in males. Up to two thirds of children with childhood absence epilepsy are girls.9,27 Absence epilepsy with myoclonus has a male predominance.28 Age The generalized idiopathic epilepsies have age-related onset. Onset of absence seizures in children with symptomatic generalized epilepsies depends on the underlying disorder. While many of these disorders may have their onset at an early (prenatal, perinatal, or postnatal) age, absence seizures do not appear until later in childhood. An example is the LennoxGastaut syndrome. The cause may be a genetic disorder or a perinatal insult, but the absence seizures do not present until age 1-8 years.29 Childhood absence epilepsy onset is at age 4-8 years, with peak onset at age 6-7 years.27 Juvenile absence epilepsy onset is generally around puberty. Actual age of onset may vary, depending on whether pyknoleptic (8.3 ± 4.5 years) or nonpyknoleptic seizures occur (14.8 ± 8.3 years).30 Juvenile myoclonic epilepsy has a more varied age of onset (8-26 y), but 79% of patients have an onset at age 12-18 years.31 Because the absence and myoclonic seizures are brief, they often go unrecognized, and many patients do not present until they experience a tonic-clonic seizure. Clinical History Children with idiopathic generalized epilepsies may present with a history of staring spells, but infrequent absence seizures may not be diagnosed until a generalized tonicclonic seizure has occurred. o Other symptoms, such as behavioral problems, may be the presenting complaint. Although the brief attacks are unrecognized, the lapses of awareness interfere with attention; as a result, the child becomes frustrated. o Decline in school performance may be an indication of the onset or breakthrough of absence seizures. In symptomatic generalized epilepsies, atypical absence seizures often occur in the setting of developmental delay or mental retardation. (See Table 1 for features of typical and atypical absence seizures.) Other seizure types can be present, such as myoclonic, tonic, atonic, tonic-clonic, and evenpartial seizures. Physical Physical and neurologic findings are normal in children with idiopathic generalized epilepsies. Having the child hyperventilate for 3-5 minutes can often provoke absence seizures. This procedure can easily be performed in the clinic or office, and the result is diagnostic. Ictal features o On clinical examination, typical absence seizures appear as brief staring spells. Patients have no warning or postictal phase, and if engaged in gross motor activity, such as walking, they may stop and stand motionless or they may continue to walk. Children are not responsive during the seizure and have no memory of what happened during the attack; they are generally unaware that a seizure has occurred. Table 1. Clinical and EEG Findings in Typical and Atypical Absence Seizures* (Adapted from Dreifuss, 1997 32 .) Open table in new window Type of Clinical Seizure EEG Findings Impairment of consciousness only Mild clonic components Atonic components Typical absence Tonic component Usually regular and symmetrical 3 Hz, possible 2- to 4-Hz spike-and-slow-wave complexes, and possible multiple spike-and-slow-wave complexes Automatisms Autonomic components Atypical absence Changes in tone more pronounced than those of typical absence seizure Nonabrupt onset or cessation abrupt EEG more heterogeneous than in typical absence; may include irregular spike-and-slow-wave complexes, fast activity, or other paroxysmal activity; abnormalities bilateral but often irregular and asymmetric *May be seen alone or in combination. o Absence seizures may be confused with complex partial seizures, especially in cases of prolonged seizures with automatisms (see Table 2). The occurrence of automatisms is dependent on duration of the seizure; the longer the seizure, the more likely automatisms are to occur (see Media file 1).33 Percentage of absence seizures with automatisms as a function of duration in seconds. (Data gathered from Penry et al, 1975 33.) o Atypical absence seizures, which occur in patients with symptomatic generalized epilepsies, are usually longer than typical absences and often have more gradual onset and resolution. o Although absence seizures may share many clinical features with complex partial seizures, the abrupt ending of typical absence seizures, without a postictal phase, is the most useful clinical feature in distinguishing the 2 conditions. Table 2. Differentiating Features of Complex Partial and Absence Seizures Open table in new window Feature Complex Partial Absence Onset May have simple partial onset Abrupt Duration Usually >30 s Usually <30 s Automatisms Present Duration dependent Awareness No No Ending Gradual postictal Abrupt In symptomatic generalized epilepsies, physical and neurologic findings may be abnormal, reflecting the underlying disorder. o o Physical examination may reveal stigmata of a genetic disease, such as a neurocutaneous disorder (eg, tuberous sclerosis) or an inborn error of metabolism. Neurologic examination may show signs of developmental delay or more specific signs, such as spastic paresis in cerebral palsy. Causes After noncompliance with treatment, lack of sleep is the most frequent cause of seizure exacerbations. Drugs that lower the seizure threshold (eg, alcohol, cocaine, high-dose penicillin, isoniazid [INH] overdose, neuroleptics) are most likely to cause seizures in patients with epilepsy. Withdrawal of alcohol, benzodiazepines, and other sedatives are also common causes. Workup Laboratory Studies When evaluating a child for staring spells, laboratory tests for metabolic abnormalities or toxic or drug ingestion (especially in older children) may be indicated. If a clear history of the episodic nature of the attacks is obtained, then the EEG can be diagnostic and laboratory tests may not be necessary. When evaluating a child with a developmental delay, or if the EEG reveals atypical absences, then a full work-up for the underlying cause of a symptomatic generalized epilepsy is indicated. Imaging Studies Neuroimaging findings are normal in idiopathic epilepsies by definition1,34 and therefore neuroimaging is not indicated if the typical clinical pattern is present. Neuroimaging is often ordered by primary care providers and the emergency department, especially if a child presents with a generalized tonic-clonic seizure, to rule out significant structural causes of seizures. A normal result helps support the diagnosis of idiopathic epilepsy. For cryptogenic and symptomatic generalized epilepsies, neuroimaging can help in diagnosing of any underlying structural abnormality. If imaging is performed, MRI is preferred to CT scanning. MRI is more sensitive for certain anatomic abnormalities. A review of 134 MRIs in patients with idiopathic generalized epilepsies found nonspecific abnormalities in 24%.35 Other Tests The only diagnostic test for absence seizures is the EEG. Findings in typical absence seizures include the following: o Background activity is normal. In syndromes with frequent absence seizures, such as childhood absence epilepsy, a routine awake recording is often pathognomonic. Bursts of frontally predominant, generalized 3-Hz spike-and-wave complexes are seen during the seizures.4 In syndromes with less frequent absence seizures (juvenile absence epilepsy or juvenile myoclonic epilepsy), an awake recording may be normal; a sleep or sleep-deprived recording may be needed. o Typical absence seizures have generalized 3-Hz spike-and-wave complexes (see Media file 2). EEG of a typical absence seizure with 3-Hz spike-and-wave discharges. The spike frequency is often faster at the onset, with a slight deceleration at the end.27 They can range from 2.5-6 Hz, with the faster frequencies seen in syndromes with older age of onset. Bursts of generalized polyspikes and waves (multiple spike-and-slow-wave complexes) may also be seen31 , especially during sleep and in syndromes with older age of onset. The onset and ending of these seizures are abrupt; no postictal EEG slowing is noted. Hyperventilation often provokes these seizures and should be a routine part of all EEGs in children. Photosensitivity may be present in idiopathic generalized epilepsies and is more often seen in juvenile myoclonic epilepsy and childhood absence epilepsy than juvenile absence epilepsy.30 EEG video monitoring demonstrates that clinical seizure manifestations may lag behind the start of ictal EEG activity; bursts lasting less than 3 seconds are usually clinically silent. During the absence seizure, rhythmic eye blinks and mild clonic jerks may be present. As a seizure progresses, automatisms may be seen.33 Clinical and EEG features may vary considerably in different children.36 Findings in atypical absence seizures include the following: o Seizures are characterized by slow spike-and-wave paroxysms, classically 2.5 Hz (see Media file 3). The onset may be difficult to discern, and postictal EEG slowing may be noted. Slow spike-and-wave discharges (2.5 Hz). This is an interictal pattern in a child with seizures and developmental delay. o o o Background activity is often abnormal, reflecting the diffuse or multifocal underlying encephalopathy of symptomatic generalized epilepsy. Generalized polyspike-and-wave complexes also may be present, and focal features may be observed. The clinical correlation of generalized spike-and-wave complexes with clinical seizures is not as clear-cut as in typical absence seizures. Generalized slow spikes and waves may be present as an interictal pattern, as in Lennox-Gastaut syndrome. o EEG-video monitoring can show a more varied alteration of consciousness than in typical absence seizures. If the patient has underlying mental retardation, discerning changes in mental status also may be more difficult in atypical absence. o Changes in postural tone, most noticeably head nods, are common. Ambulatory EEG monitoring over 24 hours may be useful to quantitate the number of seizures per day and their most likely times of occurrence. Treatment Medical Care Treatment involves antiepileptic drugs (AEDs). Once the proper diagnosis (ie, of the specific epilepsy syndrome) is made, the likelihood of other coexistent seizure types, such as myoclonic or tonic-clonic, should be considered and an appropriate medication selected. Since altered awareness occurs with even brief bursts of spike-wave paroxysms on EEG, treatment should be titrated to suppressing all epileptiform activity.37 Ethosuximide (Zarontin) is effective only against absence seizures. Valproic acid (Depakene, Depacon) and divalproex sodium (Depakote, Depakote ER) are considered broad-spectrum AEDs because they are effective against absence, myoclonic, tonic-clonic, and partial seizures. Symptomatic generalized epilepsies are often refractory to first-line AEDs. Lamotrigine (Lamictal) and topiramate (Topamax) are approved by the FDA as adjunctive therapy for the generalized seizures of Lennox-Gastaut syndrome in adult and pediatric patients (³ 2 y). Clonazepam (Klonopin, felbamate (Felbatol), topiramate, and the ketogenic or medium-chain triglyceride diet have been attempted to reduce seizure frequency. However, these adjunctive therapies have limited efficacy. Some AEDs can aggravate seizures, especially in cryptogenic or symptomatic generalized epilepsies.38Treatment with carbamazepine (Tegretol, Tegretol XR, Carbatrol)39,40 and oxcarbazepine (Trileptal)41 has been associated with the exacerbation of absence seizures. Gabapentin (Neurontin) is ineffective against absence seizures42 and tiagabine (Gabitril) and vigabatrin (Sabril) have been associated with the exacerbation of absence or myoclonic seizures in some patients.43,44 Consultations All patients with suspected absence seizures should be examined by a neurologist who has expertise in diagnosing epileptic syndromes. Patients with refractory seizures, especially those with symptomatic epilepsies, may need to be referred to an epileptologist for prolonged EEG video monitoring and medication adjustments. Diet Patients with medically intractable seizures may be tried on a ketogenic45 or medium-chain triglyceride diet46 . Although these diets are difficult to maintain, there is evidence for their effectiveness.47 Children in whom such diets are being considered should be referred to a center with specialized dietary services. Activity Physical activity should not be restricted any more than necessary. Activities in which a seizure might pose a threat, such as swimming or rock climbing, may be allowed with appropriate supervision. A child with epilepsy should not be unnecessarily handicapped. Patients with uncontrolled absence seizures should not be allowed to drive. The situation may be unclear when the patient's clinical seizures are controlled but the EEG still shows some spike-wave activity. Medication The decision to start antiepileptic medication must be made with great care. Most AEDs are relatively toxic and can have sedative and cognitive side effects. Children with absence seizures may need to be on medication for many years, and in some for life. EEG can usually confirm the diagnosis and the presence of spontaneous seizures can be documented on routine EEG or with longer recordings (ie, 24-hour ambulatory EEG or EEG video monitoring). Most AEDs are not effective against absence seizures. Also, many patients have both absence and generalized convulsive (myoclonic and generalized tonic-clonic) seizures and need an AED with efficacy for both. Only 2 first-line AEDs have FDA approval to be indicated for absence seizures: ethosuximide and valproic acid. Ethosuximide has efficacy for absence only and valproic acid has efficacy for absence, generalized tonic-clonic, and myoclonic seizures. Of the newer AEDs, lamotrigine, topiramate, and levetiracetam have been shown to have efficacy against seizures in idiopathic generalized epilepsy48,49 and have received FDA approval to be indicated for adjunctive therapy of generalized tonic-clonic seizures in idiopathic generalized epilepsy in children 2 and older (for lamotrigine and topiramate) and in children 6 and older (for levetiracetam). Lamotrigine and topiramate are also approved as adjunctive therapy in Lennox-Gastaut syndrome in children 2 years and older. Topiramate has also received FDA approval as initial monotherapy for generalized tonic-clonic seizures in children 10 years and older with idiopathic generalized epilepsy. Studies have shown these medications to have anti-absence efficacy, but the data are incomplete.50 Antiepileptics If the patient has only absence seizures, then ethosuximide (Zarontin) is an appropriate medication. This may be the case for patients with childhood absence epilepsy. Ethosuximide may also be used in conjunction with an anticonvulsive AED, such as phenytoin (Dilantin) for patients at risk of tonic-clonic seizures in whom valproic acid is contraindicated. Follow-up Further Outpatient Care Children with absence seizures should be monitored closely during titration or crossover of AEDs. The dose of the medication should be increased weekly until seizures are controlled or adverse effects develop. The aim in therapy is to control seizures completely with the minimum required amount of medication to minimize adverse effects. The therapeutic effect of valproic acid for absence seizures may lag several weeks behind reaching a therapeutic level.51 Liver function test, amylase and/or lipase, and CBC results should be monitored during drug treatment to watch for adverse reactions. Drug levels should be monitored to ensure compliance and to watch for toxic levels in patients who are too young or too developmentally disabled to articulate subjective adverse effects. Complications Absence status epilepticus may occur spontaneously, as a result of a concurrent illness, or after the administration of a drug that lowers the seizure threshold. On clinical evaluation, the patient appears to be in a dreamy state with partial responsiveness and automatisms; at times the presentation may be more subtle, with only mild encephalopathy. The diagnosis is made by EEG confirmation of generalized 3-Hz spike-and-wave complexes, although the discharges may be slower and less regular than with isolated seizures. Treatment has been intravenous benzodiazepines. In some patients, this may be replaced by or supplemented with intravenous valproic acid because intravenous benzodiazepines have been reported to produce tonic status in patients with symptomatic generalized epilepsy.52 Prognosis The prognosis for the primary generalized epilepsies depends on the particular epileptic syndrome. Because seizures, particularly generalized tonic-clonic seizures, may occur well after patients appear to achieve good control, a long seizure-free period should be achieved before discontinuation of therapy is considered. The remission rate for childhood absence epilepsy is good; 80% respond to medication. Complete remission rates vary widely, perhaps dependent on the length of follow-up. o o o Generalized tonic-clonic seizures may develop in up to 40% of children with childhood absence epilepsy.27 Persistence of seizures is more likely in those with generalized tonic-clonic seizures. Early onset of absence seizures, quick response to therapy53 , and normal EEG background are good prognostic signs. Juvenile myoclonic epilepsy carries a high risk of generalized tonic-clonic seizures. o o Despite excellent control with relatively small doses of an AED, the relapse rate is greater than 90%.54 Patients with juvenile myoclonic epilepsy generally need to be treated for life, though occasional patients achieve control with careful attention to lifestyle issues (eg, adequate sleep, abstinence from alcohol). Patient Education For excellent patient education resources, visit eMedicine's Brain and Nervous System Center. Also, see eMedicine's patient education article Epilepsy. Miscellaneous Medicolegal Pitfalls The 2 main pitfalls in treatment of absence seizures both involve incorrect diagnosis. o o On occasion, a patient without epilepsy is identified as having epilepsy. Staring spells, daydreaming, migraine equivalents, and panic and/or anxiety attacks all may be confused with nonconvulsive seizures. Certain epileptic syndromes are often undiagnosed or misdiagnosed. o Patients who present with a generalized tonic-clonic seizure are often given an AED without efficacy against absence or myoclonic seizures. Their generalized tonic-clonic seizures may be controlled, but they may have unrecognized absence or myoclonic seizures. o Patients with absence seizures may be identified as having complex partial seizures, and vice versa. This leads to incorrect treatment and an inaccurate understanding of the prognosis. Careful history taking and EEG studies can help avoid these pitfalls. Special Concerns Patients who are old enough to drive should be warned about driving and operating heavy machinery. Physicians should be familiar with state laws concerning driving with epilepsy; inform patients concerning these legal matters. Women of childbearing age who are not using adequate birth control should not be treated with valproic acid, if equally effective alternatives are available for them. o If a woman taking valproic acid wishes to become pregnant, treatment may be crossed over to ethosuximide if only absence seizures are present, and she may be given folic acid 1-5 mg/d before conception. After the first trimester, treatment may be switched back to valproic acid. o o Women with generalized tonic-clonic seizures may be crossed over to lamotrigine, and given folic acid 1-5 mg/d before conception. Most clinicians believe that women treated with valproic acid or any hepatic enzyme-inducing AED should be treated with vitamin K before delivery. Myoclonic seizures Myoclonus (pronounced /maɪˈɒklənəs/) is brief, involuntary twitching of a muscle or a group of muscles. It describes a medical sign and, generally, is not a diagnosis of a disease. The myoclonic twitches are usually caused by sudden muscle contractions; they also can result from brief lapses of contraction. Contractions are called positive myoclonus; relaxations are called negative myoclonus. The most common time for people to encounter them is while falling asleep (hypnic jerk), but myoclonic jerks are also a sign of a number of neurological disorders.Hiccups are also a kind of myoclonic jerk specifically affecting the diaphragm. Also when a spasm is caused by another person it is known as a "provoked spasm". Shuddering attacks with babys also fall in this category. Myoclonic jerks may occur alone or in sequence, in a pattern or without pattern. They may occur infrequently or many times each minute. Most often, myoclonus is one of several signs in a wide variety of nervous system disorders such as multiple sclerosis, Parkinson's disease,Alzheimer's disease, Subacute sclerosing panencephalitis and Creutzfeldt-Jakob disease (CJD) and some forms of epilepsy. Some researchers indicate that jerks persistently may even cause early tremors. In almost all instances in which myoclonus is caused by Central Nervous System (CNS) disease it is preceded by other symptoms; for instance, in CJD it is generally a late-stage clinical feature that appears after the patient has already started to exhibit gross neurological deficits. Anatomically, myoclonus may originate from lesions of the cortex, subcortex or spinal cord. The presence of myoclonus above the foramen magnum effectively excludes spinal myoclonus, but further localisation relies on further investigation with electromyography (EMG) and electroencephalography (EEG). Symptoms Myoclonic seizures can be described as "jumps." They are caused by rapid contraction and relaxation of the muscles. People without epilepsy can suffer small but similar jerks in the form of hiccups or brief twitches. These are perfectly normal. In someone with epilepsy, myoclonic seizures cause abnormal movements on both sides of the body at the same time. In reflex epilepsies, myoclonic seizures can be brought on by flashing lights or other environmental triggers (see photosensitive epilepsy). Familiar examples of normal myoclonus include hiccups and hypnic jerks that some people experience while drifting off to sleep. Severe cases of pathologic myoclonus can distort movement and severely limit a person's ability to sleep, eat, talk, and walk. Myoclonic jerks commonly occur in individuals with epilepsy. The most common types of myoclonus include action, cortical reflex, essential, palatal, progressive myoclonus epilepsy, reticular reflex, sleep, and stimulus-sensitive. Types In juvenile myoclonic epilepsy, seizures usually involve the neck, shoulders, and upper arms. These seizures typically occur shortly after waking up. They normally begin between puberty and early adulthood. They can usually be controlled with medication, but it must be taken for life. In rare cases, myoclonic seizures can be symptomatic of Lennox-Gastaut syndrome, beginning in early childhood and usually involving the face, neck, shoulders, and upper arms. In these cases, the seizures tend to be strong and difficult to control. Progressive myoclonic epilepsy includes both myoclonic and tonic-clonic seizures. Treatment is not normally successful for any extended period of time. Classifying the many different forms of myoclonus is difficult because the causes, effects, and responses to therapy vary widely. Listed below are the types most commonly described: Action myoclonus is characterized by muscular jerking triggered or intensified by voluntary movement or even the intention to move. It may be made worse by attempts at precise, coordinated movements. Action myoclonus is the most disabling form of myoclonus and can affect the arms, legs, face, and even the voice. This type of myoclonus often is caused by brain damage that results from a lack of oxygen and blood flow to the brain when breathing or heartbeat is temporarily stopped. Cortical reflex myoclonus is thought to be a type of epilepsy that originates in the cerebral cortex - the outer layer, or "gray matter," of the brain, responsible for much of the information processing that takes place in the brain. In this type of myoclonus, jerks usually involve only a few muscles in one part of the body, but jerks involving many muscles also may occur. Cortical reflex myoclonus can be intensified when patients attempt to move in a certain way or perceive a particular sensation. Essential myoclonus occurs in the absence of epilepsy or other apparent abnormalities in the brain or nerves. It can occur randomly in people with no family history, but it also can appear among members of the same family, indicating that it sometimes may be an inherited disorder. Essential myoclonus tends to be stable without increasing in severity over time. Some scientists speculate that some forms of essential myoclonus may be a type of epilepsy with no known cause. Palatal myoclonus is a regular, rhythmic contraction of one or both sides of the rear of the roof of the mouth, called the soft palate. These contractions may be accompanied by myoclonus in other muscles, including those in the face, tongue, throat, and diaphragm. The contractions are very rapid, occurring as often as 150 times a minute, and may persist during sleep. The condition usually appears in adults and can last indefinitely. People with palatal myoclonus usually regard it as a minor problem, although some occasionally complain of a "clicking" sound in the ear, a noise made as the muscles in the soft palate contract. Progressive myoclonus epilepsy (PME) is a group of diseases characterized by myoclonus, epileptic seizures, and other serious symptoms such as trouble walking or speaking. These rare disorders often get worse over time and sometimes are fatal. Studies have identified at least three forms of PME. Lafora disease is inherited as an autosomal recessive disorder, meaning that the disease occurs only when a child inherits two copies of a defective gene, one from each parent. Lafora disease is characterized by myoclonus, epileptic seizures, and dementia (progressive loss of memory and other intellectual functions). A second group of PME diseases belonging to the class of cerebral storage diseases usually involves myoclonus, visual problems, dementia, and dystonia (sustained muscle contractions that cause twisting movements or abnormal postures). Another group of PME disorders in the class of system degenerations often is accompanied by action myoclonus, seizures, and problems with balance and walking. Many of these PME diseases begin in childhood or adolescence. Reticular reflex myoclonus is thought to be a type of generalized epilepsy that originates in the brainstem, the part of the brain that connects to the spinal cord and controls vital functions such as breathing and heartbeat. Myoclonic jerks usually affect the whole body, with muscles on both sides of the body affected simultaneously. In some people, myoclonic jerks occur in only a part of the body, such as the legs, with all the muscles in that part being involved in each jerk. Reticular reflex myoclonus can be triggered by either a voluntary movement or an external stimulus. Spinal myoclonus is myoclonus originating in the spinal cord, including segmental and propriospinal myoclonus. The latter is usually due to a thoracic generator producing truncal flexion jerk. It is often stimulus-induced with a delay due to the slow conducting propriospinal nerve fibers. [1] Stimulus-sensitive myoclonus is triggered by a variety of external events, including noise, movement, and light. Surprise may increase the sensitivity of the patient. Sleep myoclonus occurs during the initial phases of sleep, especially at the moment of dropping off to sleep. Some forms appear to be stimulus-sensitive. Some persons with sleep myoclonus are rarely troubled by, or need treatment for, the condition. However, myoclonus may be a symptom in more complex and disturbing sleep disorders, such as restless legs syndrome, and may require treatment by a doctor. Cause Rarely does Myoclonus indicate anything other than arbitrary muscle contraction. Myoclonus may develop in response to infection, head or spinal cord injury, stroke, brain tumors, kidneyor liver failure, lipid storage disease, chemical or drug poisoning, as a side effect of certain drugs (e.g. tramadol[2] and quinolones), or other disorders. Benign myoclonic movements are commonly seen during the induction of general anesthesia with intravenous medications such as etomidate and propofol. These are postulated to result from decreased inhibitory signaling from cranial neurons. Prolonged oxygen deprivation to the brain, called hypoxia, may result in posthypoxic myoclonus. Myoclonus can occur by itself, but most often it is one of several symptoms associated with a wide variety of nervous system disorders. For example, myoclonic jerking may develop in patients with multiple sclerosis, Parkinson's disease, Alzheimer's disease, or Opsoclonus Myoclonus, or Creutzfeldt-Jakob disease or lupus. Myoclonic jerks commonly occur in persons with epilepsy, a disorder in which the electrical activity in the brain becomes disordered leading to seizures. It is also found in MERRF ( Myoclonic epilepsy and Red Ragged Fibres), a rare mitochondrial encephalomyopathy. Pathophysiology Although some cases of myoclonus are caused by an injury to the peripheral nerves, most myoclonus is caused by a disturbance of the central nervous system. Studies suggest that several locations in the brain are involved in myoclonus. One such location, for example, is in the brainstem close to structures that are responsible for the startle response, an automatic reaction to an unexpected stimulus involving rapid muscle contraction. The specific mechanisms underlying myoclonus are not yet fully understood. Scientists believe that some types of stimulus-sensitive myoclonus may involve overexcitability of the parts of the brain that control movement. These parts are interconnected in a series of feedback loops called motor pathways. These pathways facilitate and modulate communication between the brain and muscles. Key elements of this communication are chemicals known as neurotransmitters, which carry messages from one nerve cell, or neuron, to another. Neurotransmitters are released by neurons and attach themselves to receptors on parts of neighboring cells. Some neurotransmitters may make the receiving cell more sensitive, while others tend to make the receiving cell less sensitive. Laboratory studies suggest that an imbalance between these chemicals may underlie myoclonus. Some researchers speculate that abnormalities or deficiencies in the receptors for certain neurotransmitters may contribute to some forms of myoclonus. Receptors that appear to be related to myoclonus include those for two important inhibitory neurotransmitters: serotonin, which constricts blood vessels and brings on sleep, and gamma-aminobutyric acid (GABA), which helps the brain maintain muscle control. Other receptors with links to myoclonus include those for opiates, drugs that induce sleep, and for glycine, an inhibitory neurotransmitter that is important for the control of motor and sensory functions in the spinal cord. More research is needed to determine how these receptor abnormalities cause or contribute to myoclonus. Treatment Treatment of myoclonus focuses on medications that may help reduce symptoms. The drug of first choice to treat myoclonus, especially certain types of action myoclonus, is clonazepam, a benzodiazepine. Dosages of clonazepam usually are increased gradually until the patient improves or side effects become harmful. Drowsiness and loss of coordination are common side effects. The beneficial effects of clonazepam may diminish over time if the patient develops a tolerance for the drug. Many of the drugs used for myoclonus, such as barbiturates, phenytoin and primidone, are also used to treat epilepsy. Barbiturates slow down the central nervous system and cause tranquilizing or antiseizure effects. Phenytoin and primidone are effective antiepileptics drugs, although phenytoin can cause liver failure or have other harmful long-term effects in patients with PME. Sodium valproate is an alternative therapy for myoclonus and can be used either alone or in combination with clonazepam. Although clonazepam and/or sodium valproate are effective in the majority of patients with myoclonus, some people have adverse reactions to these drugs. Some studies have shown that doses of 5-hydroxytryptophan (5-HTP) leads to improvement in patients with some types of action myoclonus and PME. These differences in the effect of 5-HTP on patients with myoclonus have not yet been explained, but they may offer important clues to underlying abnormalities in serotonin receptors. The complex origins of myoclonus may require the use of multiple drugs for effective treatment. Although some drugs have a limited effect when used individually, they may have a greater effect when used with drugs that act on different pathways or mechanisms in the brain. By combining several of these drugs, scientists hope to achieve greater control of myoclonic symptoms. Some drugs currently being studied in different combinations include clonazepam, sodium valproate, piracetam, and primidone. Hormonal therapy also may improve responses to antimyoclonic drugs in some people. Alcohol taken before sleep seems to help in relieving the symptoms, but long-term use of alcohol is not recommended and it is not a cure. Prognosis Although myoclonus is not a life-threatening condition, it may result in serious, debilitating impairments. Action myoclonus, with its positive and negative myoclonus components, is generally considered the most serious.